硫酸氢氯吡格雷合成工艺研究
2012-01-11胡佳鹏刘志滨
胡佳鹏 卢 鑫 刘志滨
(1.浙江工业大学药学院,浙江 杭州 310032 2.浙江普洛医药科技有限公司,浙江 东阳 322118)
氯吡格雷(Clopidogre)化学名称:(S)-α-(2-氯苯基)-6,7-二氢噻吩并〔3,2-C〕吡啶-5-(4)-乙酸甲酯,商品名称:波立维(Plavix),其化学结构式如Fig1的〔1〕所示。
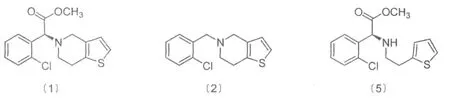
Fig.1
本品是由法国Sanofi公司于上世纪80年代中期研制开发成功的,用作抗血小板聚集药物,常以硫酸氢盐形式给药。作为血小板聚集的抑制剂,是与该公司先期开发的噻氯匹定(Ticlopidine)〔2〕同属于噻吩并吡啶的衍生物,与阿司匹林(Aspirin)广泛用于血小板聚集的抑制,在治疗心脑血管、动脉粥样硬化的血栓性疾病方面,前者比后二者更为有效和安全[1],常将〔1〕与阿司匹林联用,以达到增强疗效和降低使用者费用等目的。其作用机制是有选择性地、不可逆的抑制二磷酸腺苷(ADP)与血小板受体结合,能够阻断ADP释放后所引起的血小板活化扩增,从而达到抑制其他激动剂所诱发的血小板聚集[2]。
氯吡格雷合成方法报道的较多,但是从原料和光学拆分过程的不同进行归类,其合成方法有许多相似之处,仅仅是前后次序不同而已[3]。本论文从经济性、安全性以及环保要求等方面考虑,采用易得原料、无毒易回收溶剂,反应过程采取程序升温,达到节能减排的目的,制得氯吡格雷关键中间体〔5〕,最后一步环合得到硫酸氢氯吡格雷,合成路线图如Scheme1所示。
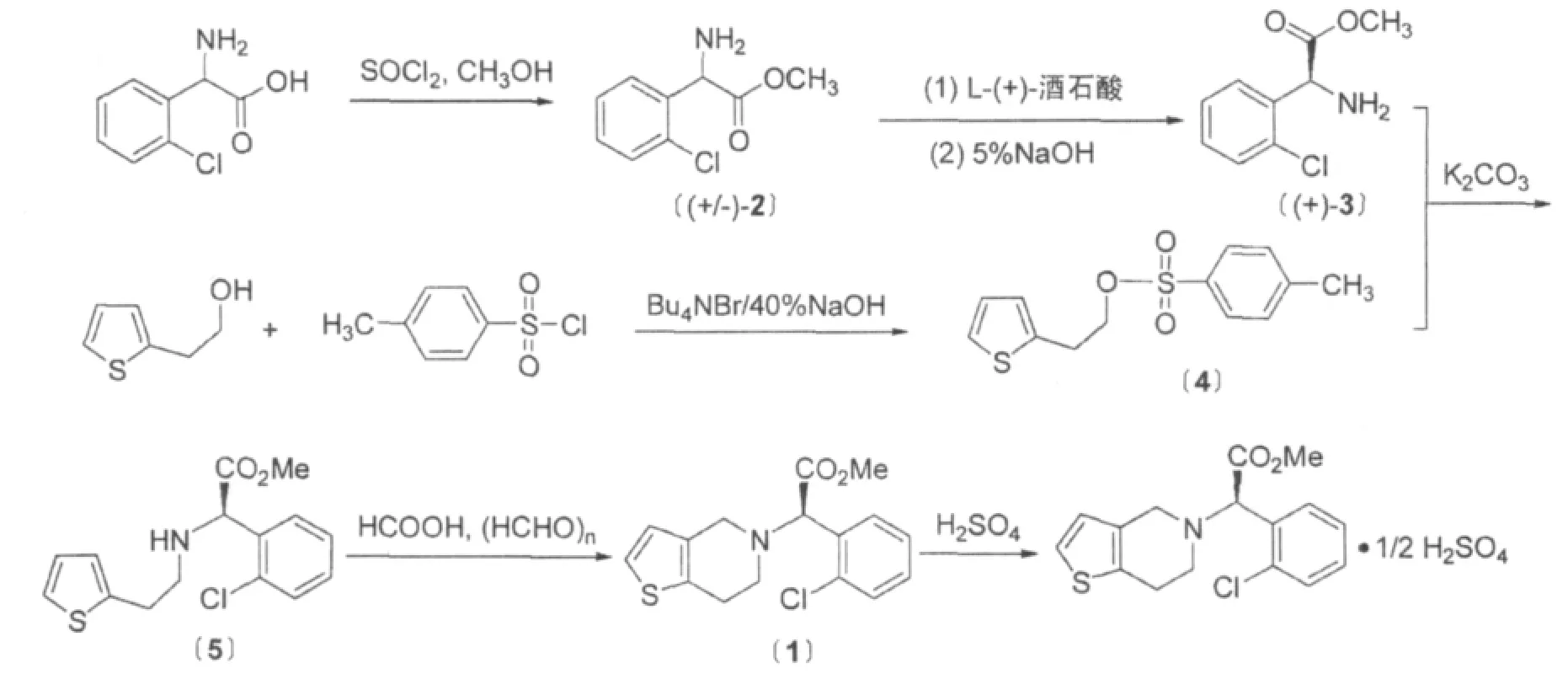
Scheme 1.硫酸氢氯吡格雷合成路线
采用(+)-邻氯苯甘氨酸为起始原料,经甲酯化得到(+)-邻氯苯甘氨酸甲酯〔2〕,经L-(+)酒石酸手性拆分得到(+)-邻氯苯甘氨酸甲酯·L-(+)酒石酸盐,然后用稀碱中和得到光学纯(+)-邻氯苯甘氨酸甲酯(+)-3。另外,噻吩乙醇和对甲苯磺酰氯反应得到对甲苯磺酸-2-噻吩乙酯〔4〕。在K2CO3作用下,化合物(+)-3和〔4〕在K2CO3催化下缩合得到(S)-2-(2-噻吩乙胺基)(2-氯苯基)乙酸甲酯,然后经Pictet-Splengler环合反应制得氯吡格雷游离碱,最后与硫酸成盐得到目标化合物硫酸氢氯吡格雷。
1 实验部分
1.1 仪器与原辅料
熔点采用WRS-1A数字显示熔点仪测定,温度未经校正。1H-NMR谱采用Bruker AV 400型核磁共振仪测定,TMS为内标。红外光谱采用FTIR-8400S型光谱仪测定。质谱采用Finigan LCQ-MS液质联用仪测定。所有试剂均为分析纯,使用前未经进一步提纯。
1.2 实验操作
1.2.1 (+)-邻氯苯甘氨酸甲酯〔(+/-)-2〕的制备
在三口烧瓶中放入100g(0.54mol)(+)-邻氯苯甘氨酸、400mL甲醇,控制温度在5℃~20℃之间滴加94g(0.80mol)氯化亚砜。滴加完毕,升温搅拌回流3h,减压浓缩蒸除甲醇。向剩余物料中加入300mL水、200mL二氯甲烷,搅拌并滴加氨水至pH=8~9,静置分层,用100mL二氯甲烷提取水相一次,提取液与有机相合并。减压蒸除二氯甲烷,得到(+)-邻氯苯甘氨酸甲酯红棕色油状产物106.5g,收率99%,HPLC色谱纯度98.9%。
1.2.2 (+)-邻氯苯甘氨酸甲酯·L-(+)酒石酸盐的制备
在三口烧瓶中放入60g(0.30mol)(+)-邻氯苯甘氨酸甲酯、240mL甲醇和68g(0.45mol)L-(+)酒石酸,搅拌,加热至60℃~65℃溶清,再缓慢降温至30℃~35℃保温8h。抽滤,湿品鼓风干燥16h,得到(+)-邻氯苯甘氨酸甲酯·L-(+)酒石酸盐白色晶体56.8g,收率54%。
母液加入30g(0.15mol)(+)-邻氯苯甘氨酸甲酯和34g(0.23mol)L-(+)酒石酸,搅拌,加热至60℃~65℃溶清,再缓慢降温至30℃~35℃保温8h。抽滤,湿品鼓风干燥16h,又获得(+)-邻氯苯甘氨酸甲酯·L-(+)酒石酸盐52.6g,收率100%,HPLC色谱纯度99.8%。母液可继续套用,总收率在80%以上。[a]=+88.1(C=1.0,CH3OH)(文献值[a]=+88(C=1.0,CH3OH))。1H NMR(500MHz,D2O)d 3.71(s,3H),4.37(s,2H),5.55(s,1H),7.31~7.48(m,4H).13C NMR(125MHz,D2O)d 56.62,56.80,75.45,130.93,131.54,132.95,133.20,134.83,136.19,171.77,178.97。
1.2.3 (+)-邻氯苯甘氨酸甲酯〔(+)-3〕的制备
在三口烧瓶中放入(+)-邻氯苯甘氨酸甲酯·L-(+)酒石酸盐50g(0.14mol)、150mL甲苯,搅拌,降温至0℃~10℃滴加5%NaOH水溶液至pH=9,静置分层,用100mL甲苯提取水相一次,提取液与机相合并。减压蒸除甲苯,得到(+)-邻氯苯甘氨酸甲酯棕黄色油状物28g,收率98%,HPLC色谱纯度99.6%。
1.2.4 2-(2-噻吩)乙醇对甲苯磺酸酯〔4〕的制备
在三口烧瓶中放入97g(0.51mol)对甲苯磺酰氯、180mL甲苯,搅拌至溶清,过滤,并保持在15℃~20℃待用。在三口烧瓶中放入60g(0.47mol)2-噻吩乙醇、60mL甲苯、0.6g四丁基溴化铵(Bu4NBr)、120g 40%NaOH水溶液,搅拌,控制温度在10℃~20℃滴加97g(0.51mol)对甲苯磺酰氯溶于180mL甲苯的溶液。滴加完毕,继续搅拌3h,然后温升至45℃~55℃搅拌8h,静置分层,有机相减压蒸除甲苯,得到2-(2-噻吩)乙醇对甲苯磺酸酯红棕色油状物130g,收率98%,HPLC色谱纯度99.9%。1H NMR(500MHz,CDCl3)d 2.43(s,3H),3.16(t,2H,J=7.0Hz),4.21(t,2H,J=7.0Hz),6.79~6.90(m,3H),7.30(d,2H,J=8.5Hz),7.73(d,2H,J=8.5Hz).13C NMR(125MHz,CDCl3)d 21.55,29.45,70.03,124.28,126.00,126.92,127.79,129.78,132.82,137.97,144.76.
1.2.5 (S)-2-(2-噻吩乙胺基)(2-氯苯基)乙酸甲酯盐酸盐〔5〕的制备
在三口烧瓶中放入(+)-邻氯苯甘氨酸甲酯28g(0.14mol)、2-(2-噻吩)乙醇对甲苯磺酸酯48g(0.17mol)、碳酸钾29g(0.21mol),搅拌,升温至78℃~82℃反应12h,再升温至100℃~106℃反应30h。降温过滤,滤液在搅拌下滴加36%浓盐酸至pH=1~2,抽滤,湿品鼓风干燥16h,得到(S)-2-(2-噻吩乙胺基)(2-氯苯基)乙酸甲酯盐酸盐白色固体39g,收率81%,HPLC色谱纯度99.8%。mp:181℃~182℃,[a]=108.4(C=1.0,CH3OH)(文献值[a]=+108~109(C=1.0,CH3OH))。
1.2.6 (S)-(+)-α-(2-氯苯基)-6,7-二氢噻吩并[3,2-c]吡啶-5(4H)乙酸甲酯硫酸氢盐的制备
在三口烧瓶中放入45g(0.14mol)(S)-2-(2-噻吩乙胺基)(2-氯苯基)乙酸甲酯盐酸盐、160mL二氯甲烷,搅拌,滴加8℃碳酸氢钠溶液中和pH=7~8,静置分层,用80mL二氯甲烷提取水相一次,提取液与有机相合并,减压蒸除二氯甲烷待用。另一个三口烧瓶中放入7.8g多聚甲醛、210g无水甲酸,搅拌,升温至70℃~75℃溶清,降温至0~5℃。控制温度0~5℃滴加上述制备的游离碱,再升温至20℃~25℃反应5h。倒入720g冰水中,用200mL×2二氯甲烷萃取,合并有机相。滴加8%碳酸氢钠溶液中和pH=7~8,静置分层,减压蒸除二氯甲烷。控制温度0~5℃滴加10%的硫酸丙酮溶液,抽滤,得(S)-(+)-α-(2-氯苯基)-6,7-二氢噻吩并[3,2-c]吡啶-5(4H)乙酸甲酯硫酸氢盐49g,收率84%。IR(KBr)3406,3373,3009,2955,1751,1437,1477,1223,1192,1055,1076cm-1.1H NMR(500 MHz,CDCl3)d 3.10(s,2H),3.52(brs,2H),3.77(s,3H),4.28(brs,2H),5.66(s,1H),6.90~7.71(m,6H),8.77(brs,2H).13C NMR(125 MHz,CDCl3)d 22.35,49.22,50.46,53.83,65.57,125.23,125.60,127.90,128.39,128.65,130.41,130.76,131.72,132.45,134.40,167.40.
2 结论
本论文第二步拆分选用L-(+)酒石酸做拆分剂,边消旋边拆分的方法,实现母液的套用,拆分收率达到80%以上,大大提高了拆分效率,降低了生产成本。第四步缩合用甲苯代替乙腈[4-5],不仅减少了环境污染,而且降低了成本;另外,化合物〔3〕和〔4〕的缩合反应采用弱碱K2CO3为催化剂,并且采用程序升温的方法,有效地避免了产品的消旋,杂质也得到了很好的控制,提高了产品的质量。本文开发了一种氯吡格雷的合成新工艺,以消旋的邻氯苯甘氨酸为原料,经过5步反应,以52.8%的总收率得到硫酸氢氯吡格雷,该方法具有原料价廉易得、步骤简短、反应收率高、生产成本低且环境友好的优点,具有较好的工业化应用前景。
[1]贾国强,等.新型的ADP受体拮抗剂硫酸氢氯吡格雷[J].药物与临床,2002,17(5):52-53.
[2]吴荣辉.氯吡格雷联合阿司匹林与单用阿司匹林治疗急性心肌梗死的临床疗效比较[J].中国实用医学,2010,26:10-12.
[3]陈子明,陈子明,杜玉民,等.氯吡格雷合成路线图解[J].中国医药工业杂志,2002,23(4):206-208.
[4]徐斌,等.氯吡格雷及其盐的制备方法:中国,101333223 B[P].2008-12-31.
[5]吴范宏,等.氯吡格雷中间体(S)-2-(2-噻吩乙胺基)(2-氯苯基)乙酸甲酯及其盐的制备:中国,101519401 A[P].2009-09-02.
