新型4-取代四氢咪唑烷的合成*
2011-11-26高彬恒
高彬恒, 陈 垦, 徐 亮
(四川大学 华西药学院,四川 成都 610041)
四氢咪唑烷是生物活性化合物的重要结构单元[1~3],常利用邻二胺与羰基化合物的缩合反应合成[4],但是该方法受二胺化合物来源的限制。亚甲胺基叶立德与亚胺通过1,3-偶极环加成反应构建四氢咪唑烷[5,6]也是重要的合成方法,该方法利用含酯基取代的稳定型亚甲胺基叶立德,反应条件比较苛刻,产物为4,5-双取代的四氢咪唑烷。文献[7,8]方法以N-甲氧基-N-三甲硅甲基苄胺(1)作为一种非稳定型亚甲胺基叶立德前体,在氟离子、质子酸或路易斯酸的作用下与烯烃或羰基化合物反应,生成各种五元含氮杂环。
本文以三氟醋酸(TFA)催化1生成非稳定型亚甲胺基叶立德;再与N-Ts醛亚胺(2a~2j)完成1,3-偶极环加成反应合成了10个新的4-取代四氢咪唑烷(3a~3j, Scheme 1),收率80%~99%,其结构经1H NMR和13C NMR表征。
1 实验部分
1.1 仪器与试剂
Varian Unity NOVA-400/54型核磁共振仪(CDCl3为溶剂,TMS为内标)。
柱层析用硅胶,200目~300目,青岛海洋化工厂;薄层层析GF254硅胶板,烟台江友硅胶开发有限公司;其余所用试剂均为分析纯。
1.2 合成
(1)1的合成[9]
在反应瓶中加入氯甲基三甲基硅烷4.08 g(33 mmol)和CH3CN 35 mL,搅拌下滴加苄胺7.17 g(67 mmol),回流反应16 h。冷却至室温,过滤,滤液浓缩后加水50 mL,用正己烷(3×30 mL)萃取,合并萃取液,用饱和食盐水洗涤,无水硫酸钠干燥,减压蒸干得淡黄色液体A 5.8 g。
Scheme1
在烧瓶中加入37%甲醛溶液2.8 mL(36 mmol)和甲醇1.45 mL(36 mmol),冰浴冷却,搅拌下缓慢滴加A,滴毕,于0 ℃反应1 h;于10 ℃~15 ℃反应2 h;加入无水碳酸钾3.17 g(23 mmol),于室温反应2.5 h。静置分层,在上层清液中加入无水碳酸钾300 mg,搅拌20 min后过滤,滤液抽干得无色透明液体16.30 g,收率81%。
(2)3的合成(以3a为例)
氩气保护,在反应管(5 mL)中加入2a0.5 mmol,10.75 mmol和无水CH2Cl2(1.5 mL),搅拌下滴加三氟醋酸(TFA) 0.05 mmol,于室温反应12 h(TLC检测)。反应液经硅胶柱层析[洗脱剂:V(石油醚) ∶V(乙酸乙酯)=15 ∶1]纯化得白色固体3a。
用类似方法合成白色固体3b~3j。
3a:1H NMRδ: 7.60(dd,J=1.6 Hz, 6.4 Hz, 2H), 7.33~7.21(m, 10H), 7.13 (dd,J=1.6 Hz, 7.6 Hz, 2H), 4.74(t,J=7.6 Hz, 1H), 4.42(d,J=7.6 Hz, 1H), 4.08(d,J=8.0 Hz, 1H), 3.58(d,J=13.2 Hz, 1H), 3.40(d,J=13.2 Hz, 1H), 3.26(dd,J=7.6 Hz, 10.0 Hz, 1H), 2.66(dd,J=7.6 Hz, 10.4 Hz, 1H), 2.43(s, 3H);13C NMRδ: 143.5, 140.6, 137.6, 135.2, 129.5, 128.5, 128.5, 128.4, 127.8, 127.5, 127.5, 126.6, 71.4, 62.4, 62.0, 57.1, 21.6。
3b:1H NMRδ: 7.61(d,J=8.0 Hz, 2H), 7.32~7.25(m, 7H), 7.17~7.15(m, 2H), 6.83(dd,J=2.0 Hz, 6.8 Hz, 2H), 4.70(t,J=7.2 Hz, 1H), 4.40(d,J=8.0 Hz, 1H), 4.08(d,J=8.0 Hz, 1H), 3.80(s, 3H), 3.59(d,J=13.2 Hz, 1H), 3.42(d,J=13.2 Hz, 1H), 3.23(dd,J=7.2 Hz, 10.0 Hz, 1H), 2.66(dd,J=8.0 Hz, 10.0 Hz, 1H), 2.45(s, 3H);13C NMRδ: 159.0, 143.4, 137.6, 135.2, 132.7, 129.5, 128.5, 128.4, 127.8, 127.8, 127.5, 113.9, 71.3, 62.4, 61.5, 57.1, 55.3, 21.6。
3c:1H NMRδ: 7.61(dd,J=2.0 Hz, 8.4 Hz, 2H), 7.30~7.20(m, 7H), 7.14~7.08(m, 4H), 4.69(t,J=7.2 Hz, 1H), 4.40(d,J=8.0 Hz, 1H), 4.06(d,J=7.6 Hz, 1H), 3.56(d,J=12.8 Hz, 1H), 3.38(d,J=13.2 Hz, 1H), 3.23(m, 1H), 2.63(dd,J=7.6 Hz, 10.4 Hz, 1H), 2.44(s, 3H), 2.32(s, 3H);13C NMRδ: 143.5, 137.6, 137.2, 135.2, 129.5, 129.2, 128.5, 128.4, 127.8, 127.5, 126.5, 71.4, 62.4, 61.8, 57.1, 21.6, 21.1。
3d:1H NMRδ: 7.70(d,J=8.4 Hz, 2H), 7.63(dd,J=1.6 Hz, 8.0 Hz, 1H), 7.47(dd,J=1.2 Hz, 8.0 Hz, 1H), 7.32~7.24(m, 6H), 7.12~7.08(m, 3H), 5.08(t,J=7.2 Hz, 1H), 4.37(d,J=8.0 Hz, 1H), 4.13(d,J=7.6 Hz, 1H), 3.53(d,J=13.2 Hz, 1H), 3.40(dd,J=6.8 Hz, 10.0 Hz, 1H), 3.31(d,J=13.2 Hz, 1H), 2.51(dd,J=7.2 Hz, 10.4 Hz, 1H), 2.45(s, 3H);13C NMRδ: 143.9, 140.0, 137.5, 134.2, 132.5, 129.7, 128.9, 128.4, 128.1, 127.8, 127.5, 122.0, 71.6, 61.5, 60.8, 57.1, 21.6。
3e:1H NMRδ: 7.69(d,J=8.4 Hz, 2H), 7.64(dd,J=1.6 Hz, 7.6 Hz, 1H), 7.31~7.23(m, 7H), 7.17(dd,J=1.6 Hz, 7.6 Hz, 1H), 7.08(dd,J=2.0 Hz, 7.2 Hz, 2H), 5.11(t,J=7.2 Hz, 1H), 4.36(d,J=8.4 Hz, 1H), 4.10(d,J=8.0 Hz, 1H), 3.52(d,J=13.2 Hz, 1H), 3.39(dd,J=7.2 Hz, 10.4 Hz, 1H), 3.31(d,J=13.2 Hz, 1H), 2.51(dd,J=7.6 Hz, 10.4 Hz, 1H), 2.45(s, 3H);13C NMRδ: 143.8, 138.5, 137.5, 134.4, 131.9, 129.7, 129.7, 129.3, 128.6, 128.4, 128.2, 128.0, 127.5, 127.2, 71.4, 60.7, 59.3, 57.1, 21.6。
3f:1H NMRδ: 7.60(d,J=7.6 Hz, 2H), 7.30~7.25(m, 9H), 7.13(dd,J=2.0 Hz, 7.2 Hz, 2H), 4.69(t,J=7.2 Hz, 1H), 4.37(d,J=8.0 Hz, 1H), 4.08(d,J=7.6 Hz, 1H), 3.57(d,J=13.2 Hz, 1H), 3.40(d,J=12.8 Hz, 1H), 3.22(dd,J=6.8 Hz, 10.0 Hz, 1H), 2.62(dd,J=7.6 Hz, 10.4 Hz, 1H), 2.45 (s, 3H);13C NMRδ: 143.8, 139.2, 137.4, 134.9, 133.3, 129.6, 128.6, 128.5, 128.4, 128.0, 127.8, 127.5, 71.3, 62.1, 61.4, 57.0, 21.6。
3g:1H NMRδ: 7.57(d,J=8.0 Hz, 2H), 7.31~7.17(m, 9H), 6.99(d,J=3.2 Hz, 1H), 6.88(dd,J=3.2 Hz, 4.8 Hz, 1H), 5.11(t,J=6.4 Hz, 1H), 4.26(d,J=8.4 Hz, 1H), 4.09(d,J=7.2 Hz, 1H), 3.65(d,J=13.2 Hz, 1H), 3.49(d,J=13.2 Hz, 1H), 3.18(dd,J=6.8 Hz, 10.0 Hz, 1H), 2.85(dd,J=6.4 Hz, 10.4 Hz, 1H), 2.42(s, 3H);13C NMRδ: 144.4, 143.5, 137.5, 135.5, 129.5, 128.5, 128.4, 127.6, 127.5, 126.4, 125.6, 125.3, 70.3, 61.8, 57.7, 57.0, 21.6。
3h:1H NMRδ: 7.61~7.59(m, 2H), 7.31~7.26(m, 6H), 7.13(dd,J=1.6 Hz, 7.2 Hz, 2H), 6.99~6.95(m, 2H), 4.72(t,J=7.2 Hz, 1H), 4.38(d,J=7.6 Hz, 1H), 4.08(d,J=7.6 Hz, 1H), 3.57(d,J=12.8 Hz, 1H), 3.41(d,J=12.8 Hz, 1H), 3.22(m, 1H), 2.63(dd,J=7.2 Hz, 10.0 Hz, 1H), 2.45(s, 3H);13C NMRδ: 163.4, 161.0, 143.7, 137.5, 136.5, 136.5, 135.0, 129.6, 128.5, 128.3, 128.2, 127.8, 127.5, 115.4, 115.2, 71.3, 62.3, 61.4, 57.0, 21.6。
3i:1H NMRδ: 7.61~7.58(m, 2H), 7.32~7.25(m, 6H), 7.25~7.17(m, 2H), 6.31~6.27(m, 2H), 4.83(t,J=7.6 Hz, 1H), 4.40(d,J=8.0 Hz, 1H), 4.06(d,J=8.4 Hz, 1H), 3.65(d,J=12.8 Hz, 1H), 3.48(d,J=12.8 Hz, 1H), 3.20(m, 1H), 2.98(dd,J=8.0 Hz, 10.8 Hz, 1H), 2.43(s, 3H);13C NMRδ: 152.5, 143.4, 142.2, 137.7, 135.8, 129.5, 128.6, 128.4, 127.6, 127.5, 110.4, 108.1, 70.7, 58.2, 57.2, 55.0, 21.5。
3j:1H NMRδ: 7.70(d,J=8.4 Hz, 2H), 7.32(d,J=8.0 Hz, 2H), 7.29~7.23 (m, 3H), 7.01~6.99(m, 2H), 4.20(d,J=8.4 Hz, 1H), 3.64(m, 1H), 3.63(d,J=8.0 Hz, 1H), 3.47(d,J=13.2 Hz, 1H), 3.28(d,J=13.2 Hz, 1H), 2.77(dd,J=7.6 Hz, 9.6 Hz, 1H), 2.48(s, 3H), 2.32(dd,J=7.6 Hz, 9.6 Hz, 1H), 2.20~2.15(m, 1H), 0.95(d,J=6.8 Hz, 3H), 0.91(d,J=6.8 Hz, 3H);13C NMRδ: 143.4, 137.8, 135.2, 129.5, 128.3, 128.2, 128.0, 127.3, 70.8, 64.3, 56.8, 53.7, 31.5, 21.6, 19.3, 16.6。
2 结果与讨论
2.1 催化剂的筛选
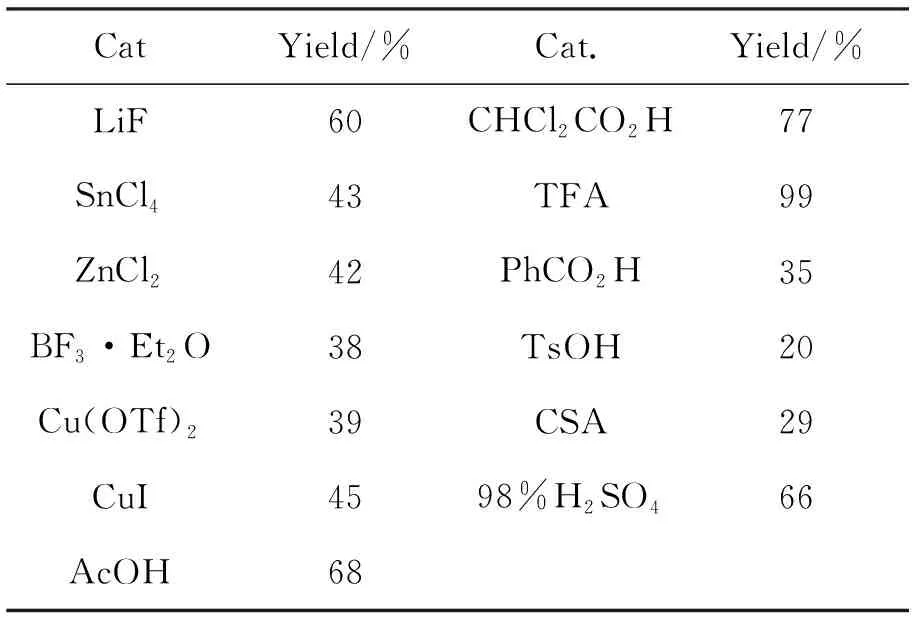
表 1 催化剂的筛选*
*催化剂10 mol%,其余反应条件同1.2(2)
以2a作底物,10 mol%催化剂,其余反应条件同1.2(2),对催化剂进行筛选,结果见表1。从表1可以看出,各种类型的催化剂均能不同程度地催化反应发生,得到预期的环加成产物3a。文献报道氟化锂催化1与各种贫电子烯烃发生环加成反应,但是在本反应的筛选中,仅获得60%的收率。路易斯酸也可以催化反应,但是活性较低。质子酸的催化活性相对高,其中TFA几乎达到了定量收率,是最佳催化剂。无机质子酸硫酸也可以获得较好的收率。磺酸类质子酸表现不佳,收率低于30%。
2.2 亚胺底物的拓展
以TFA(10 mol%)为催化剂,其余反应条件同1.2(2),拓展底物(2a~2j)的实验结果见表2。从表2可以看出,1能与2发生1,3-偶极环加成反应,生成相应的3。无论是富电子的还是贫电子的取代苯基对甲苯磺酰亚胺(2a~2g),或是芳杂醛对甲苯磺酰亚胺(2h~2i),均获得了90%以上的高收率。异丁醛亚胺(2j)也可以在此条件下获得良好的收率(80%),这说明该反应对脂肪醛亚胺也具有良好的底物适应性。
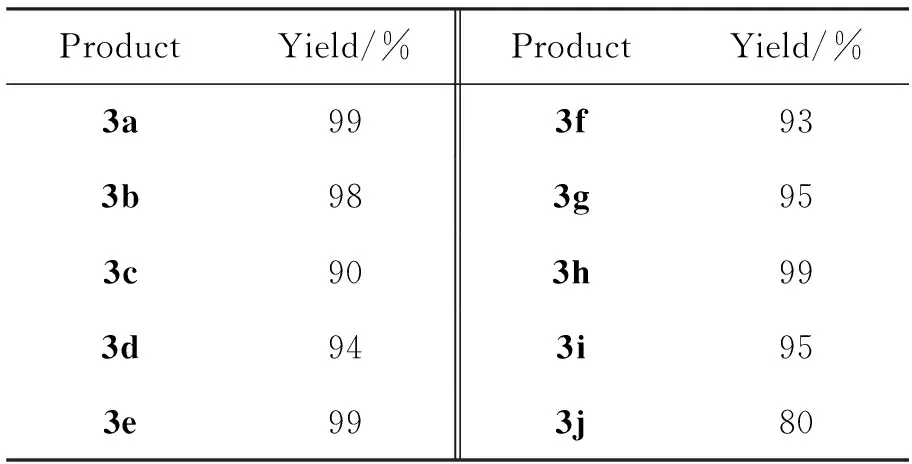
表 2 亚胺底物的拓展*
*以TFA(10 mol%)为催化剂,其余反应条件同1.2(2)
[1] Sharma V, Crankshaw C L, Piwnica-Worms D. Effects of multidrug resistance(MDR1)p-glycoprotein expression levels and coordination metal on the cytotoxic potency of multidentate(N4O2)(Ethylenediamine) bis[propyl(R-benzylimino)]metal(Ⅲ) Cations[J].J Med Chem,1996,39:3483-3490.
[2] Sage C R, Michelitsch M D, Stout T J,etal. D221 in thymidylate synthase controls conformation change,and thereby opening of the imidazolidine[J].Biochemistry,1998,37:13893-13901.
[3] Chang-Fong G, Benamour K, Szymonki B,etal. Synthesis andα-adrenergic binding ligand affinities of 2-imino-midazolidine derivatives[J].Chem Pharm Bull,2000,48(5)729-733.
[4] Katritzky A, Suzuki K, He Hai-Ying. Convenient syntheses of unsymmetrical imidazolidines[J].J Org Chem,2002,67:3109-3114.
[5] Viso A, de la Pradilla F, Garcia A,etal. Highly diastereoselective[3+2] cycloadditions between nonracemicp-tolylsulfinimines and iminoesters:An efficient entry to enantiopureImidazolidines and vicinal diaminoalcohols[J].Chem Eur J,2003,9:2867-2876.
[6] Padwa A, Dean D, Osterhout M,etal. Synthesis of function alized azomethine ylides via the Rh(Ⅱ)-catalyzed cyclization ofalpha-diazo carbonyls onto iminopi-bonds[J].J Org Chem,1994,59:5347-5357.
[7] Terao Y, Kotaki H, Imai N,etal. Trifluoroacetic acid-catalyzed 1,3-cycloaddition of the simplest iminium ylide leading to 3- or 3,4-substituted pyrrolidines and 2,5-dihydropyrroles[J].Chem Pharm Bull,1985,33:2762-2766.
[8] Padwa A, Dent W. On the use ofN-[(trimethylsilyl)methyl]amino ethers as capped azomethine ylide equivalents[J].J Org Chem,1987,52:235-244.
[9] Kotian P, Lin T, El-Kattan,etal. A practical large-scale synthesis of (3R,4R)-4-(hydroxymethyl)pyrrolidin-3-olvia asymmetric 1,3-dipolar cycloaddition[J].Organic Process Research & Development,2005,9:193-197.
