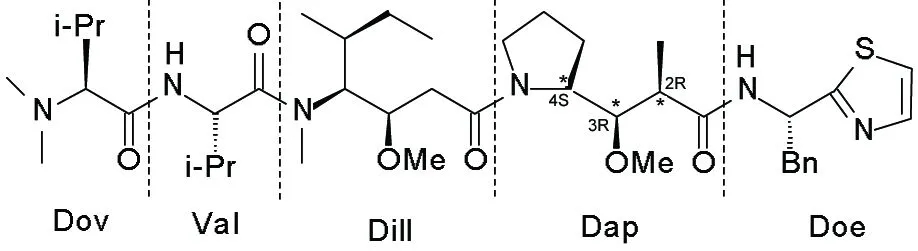
海兔毒素10关键合成子Dap的立体专一性合成*
2011-11-24梁兴勇邓丽敏杨劲松
高 亮, 梁兴勇, 邓丽敏, 杨劲松
(四川大学 华西药学院,四川 成都 610041)
海兔毒素10(D-10, Chart 1)是1987年美国Pettit小组从西印度洋软体动物海兔中分离得到一种高活性的抗癌活性肽,主要用于小细胞肺癌、卵巢癌、黑素瘤和前列腺等实体瘤的治疗,目前已进入Ⅱ期临床[1]。由于具有确切的作用机制及温和的毒性反应,D-10很有可能成为联合用药的首选[2~4]药物之一。
天然的D-10产量极小,而现有的合成方法和路线仅限于实验室规模,无法满足日益增长的需求。其根本原因在于无法高效立体选择性在合成D-10中最复杂的组分,即具有三个手性中心的Dolaproine(Dap)。传统合成Dap的方法主要有:(1)应用Reformatsky反应进行立体选择性的合成β-羟基羰基化合物[5],通过引入空间位阻大的基团来控制2′-和3′-位手性[6],或者利用Zr, Rh[7]等金属以及镧系元素,考察产物立体选择性,但结果均不理想,并且得到的四个异构体分离困难。(2)应用Baylis-Hillman反应[8]和Ru的催化作用[9],但只能单独控制2′-位甲基构型,且仍是得到一对非对映异构体。(3)Walker等[10~12]应用Evans羟醛缩合反应进行了顺式“非Evans”体研究,通过TiCl4, SnCl4等路易斯酸,利用金属螯合作用控制手性,但仍不能得到单一的顺式产物。
D-10


Scheme1
本文在以上文献方法的基础上,设计以N-(叔丁氧羰基)-S-脯氨醛(4)和N-丙酰基-(3R,4S)-3-甲基-4-苯基-2-羰基噁唑啉(5)为原料,经羟醛缩合反应制备了高立体专一性、具有三个手性中心的(4S,5S,2′R,3′R,2″S)-3-[3′-(N-叔丁氧羰基-2″-吡咯烷基)-3′-羟基-2′-甲基丙基)]-4-甲基-5-苯基-2-羰基噁唑烷酮(6); 6脱去手性辅基,甲基化3′-位羟基合成了D-10的关键合成子——(2S,3S,2′S)-3-(N-叔丁氧羰基-2′-吡咯烷基)-3-甲氧基-2-甲酸丙酸(8, Scheme 1),其结构经1H NMR,13C NMR, IR和MS表征。
改进方法具有如下优点:(1)不需使用Ti, Sn等路易斯酸,通过一步反应便可制备三个手性中心的关键中间体6; (2)通过反应条件的优化,避免了难分离异构体的生成,产物单一,不仅具有高立体选择性,同时具有高对映选择性;(3)合成路线具有反应条件温和、操作安全、后处理方便、产率高、且手性辅基可回收利用,降低成本等特点,极大简化了Dap的合成,具有工业化大规模生产前景。
1 实验部分
1.1 仪器与试剂
PE 314型自动旋光仪;Varian INOVA 400型核磁共振仪(CDCl3为溶剂,TMS为内标)。
所用试剂均为化学纯;反应均在氮气保护下进行;反应容器和试剂经干燥处理。
1.2 合成
(1)3的合成
在反应瓶中加入1 145 g(700 mmol)的THF(1 L)溶液,搅拌下于0 ℃缓慢滴加1 mol·L-1硼烷的四氢呋喃溶液 (1.35 L),滴毕,于0 ℃反应2 h;于室温反应1 h(TLC监测)。加水(2 L)终止反应,用乙酸乙酯(1.5 L)萃取,合并有机相,依次用饱和碳酸钠、水和饱和食盐水洗涤,无水硫酸钠干燥,浓缩得无色液状液体3,收率85%。
(2)4的合成
(3)5的合成
在反应瓶中加入2 100 g(560 mmol)的THF(1.60 L)溶液,搅拌下于-75 ℃缓慢滴加2.06 mol·L-1丁基锂的己烷溶液(270 mL),滴毕,加入丙酰氯55 mL,于-75 ℃反应30 min;于-60 ℃反应2 h(TLC监测)。加入饱和氯化铵溶液(400 mL)终止反应,浓缩后用二氯甲烷萃取。合并萃取液,依次用1 mol·L-1NaOH溶液、饱和食盐水洗涤,无水硫酸钠干燥,浓缩得淡黄色油状液体5,收率95%;1H NMRδ: 7.47~7.38(m, 5H, PhH), 5.85(d, 1H, CHPh), 4.84~4.77(m, 1H, CHCH3), 2.90~2.84(m, 2H, CH2), 1.06(t, 3H, CH2CH3), 0.76(d, 3H, CHCH3)。
(4)6的合成
(5) 7的合成
在反应瓶中加入6103 g(240 mmol)和THF/H2O(1.35 L/0.33 L),搅拌下于3 ℃加入30%H2O297 mL, LiOH 18.5 g(770 mmol)的水(500 mL)溶液。冰浴冷却下反应5 h后加入Na2SO3120 g(950 mmol)的水(700 mL)溶液,于室温反应16 h(TLC监测)。用二氯甲烷洗涤,二氯甲烷浓缩回收原料2;水层用0.44 mol·L-1HCl中和至pH 3后用乙酸乙萃取,合并有机层,浓缩后溶于5%NaHCO3;水层用二氯甲烷洗涤后用3 mol·L-1HCl调至pH 3;水层用乙酸乙酯萃取,合并有机层,用无水硫酸钠干燥层,浓缩得无色油状液体7,收率100%;1H NMRδ: 4.02~3.86(m, 1H, 6-H), 3.84~3.76(m, 1H, NCH), 3.57~3.50(m, 1H, 5a-H), 3.27~3.40(m, 1H, 5b-H), 2.56~2.52(m, 1H, 2′-H), 1.96~1.72(m, 4H, 3,4-H), 1.48(s, 9H, CH3in Boc), 1.28(d, 3H, CHCH3)。
(6) 8的合成
Scheme2
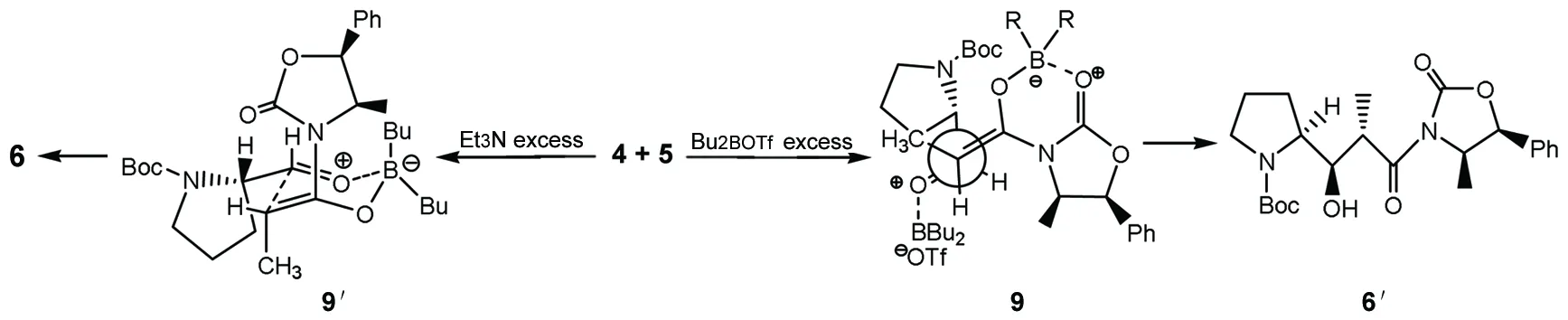
Scheme 3
Scheme4
2 结果与讨论
2.1 4的合成方法改进及1H NMR分析
用文献[6]方法(SO3·Py, Et3N, DMS)合成4时,操作繁琐,反应温度难以掌控(15 ℃~17 ℃反应40 min, 7 ℃~8 ℃反应2.5 h),产率低(28%),且产物纯化困难。实验中,我们尝试用PCC作氧化剂,室温下即可完成反应,产率提高至60%,且产物纯化简单,只需过硅胶短柱即可。
4的1H NMR显示有两套信号峰值,是因为酰胺键具有假双键的性质,造成同一化合物异化为两个旋转异构体(Scheme 2)。旋转异构体的比例与温度有关。当温度升至55 ℃[6]时,双信号合并。
2.2 6的合成及机理探讨
Et3N和Bu2BOTf的用量及加料顺序是立体选择性合成6的重要影响因素。当(Bu)2BOTf过量时,主要生成反式产物6′(6′,5和6的收率分别为53%, 38%和9%);当Et3N略过量于Bu2BOTf时,反应以生成顺式产物6为主(6和6′的收率分别为75%, 5%)。文献[15]先加入Bu2BOTf再加入Et3N,反应则以生成非对映异构体6′为主(6和6′的收率分别为75%, 4%)。我们首先加入1.44 eq.Et3N,再加入1.25 eq Bu2BOTf,只得到6,体系干净,只需过硅胶短柱滤除色素,产率78%。我们推测其可能的反应机理如Scheme 3所示。当Bu2BOTf过量时,4与过量的Bu2BOTf形成开放而非闭合的过渡态9′,最终形成Evans型反式产物9′;当Et3N略过量时,过量的碱与未反应的路易斯酸结合,4只能和噁唑烷酮5与Bu2BOTf形成的中间体偶联,而苯基与甲基的存在阻碍了4向另一面进攻,因此脯氨酸环占据空间位阻较小的e键形成过渡态9,进而得到目标产物6。
2.3 2的合成改进
用文献[16]方法由7合成8时,会生成两个较难分离的副产物11和12(Scheme 4),产率分别为5.6%和5.3%)。我们发现温度稍高会产生一不容易分离的荧光杂质;温度太低则反应不易进行,并会产生一无法分离的杂质,所以严格控制0 ℃这一反应温度是提高8产率的关键。同时我们通过加大碘甲烷的用量(由13.2 eq.提高到18.0 eq.),使得8的产率由最初的33%提高到65%,副产物仅有一个,且产率低于3%。
[1] Pettit G R, Yoshiaki K, Cherry L H,etal. Isolation of dolastatins 10~15 from the marine mollusc dolabella auricularia[J].Tetrahedron,1993,49:9151-9170.
[2] 闫世凤,王小兵. 海洋抗癌活性物质的研究进展[J].国外医学药学分册,2005,32(4):233-237.
[3] Bai R, Covell D G, Taylor G F,etal. Direct photoaffinity labeling by dolastatin 10 of the amino terminal peptide of beta tubulin containing cysteine 12[J].J Bio Chem,2004,279(29):30731-30740.
[4] Hoffman M A, Blessing J A, Lentz S S. A phase Ⅱ trial of dolastatin 10 in recurrent platinum sensitive ovarian carcinoma:A gynecologic on cology group study[J].Gynecol Oncol,2003,89(1):95-98.
[5] Hidenori D, Marvin M H, Clayton H H. Acyclic stereoselection;48:Reversal of stereochemistry in the aldol reactions of a chiral boron enolate[J].J Org Chem,1990,55(1):173-181.
[6] Pettit G R, Singh S B, Herald D L,etal. The dolastatins;17:Synthesis of dolaproine and related diastereoisomers[J].J Org Chem,1994,59(21):6287-6295.
[7] Nakamura E, Kuwajima I. [4+2]-cycloaddition of singlet oxygen to conjugated acyclic hexadienes:Evidence of singlet oxygen inducedcistrans-isomerization[J].Tetrahedron Lett,1983,24:3303-3306.
[8] Wanda P, Almeidaa B. An easy and stereoselective syn thesis ofN-Boc-dolaproine via the Baylis-Hillman reaction[J].Tetrahedron Lett,2003,44:937-940.
[9] Damien L, Céline M. Stereoselective synthesis of iso-dolaproine via dynamic kinetic resolution[J].Org Lett,2001,3(12):1909-1912.
[10] Céline M, Sébastien R, Hitoshi T,etal. Total synthesis of dolastatin 10 through ruthenium-catalyzed asymmetric hydrogenations[J].Tetrahedron,2007,63:6115-6123.
[11] Evans D A, Baetroli, J. Enantioselective aldol condensations.2.Erythro-selective chiral aldo condensations via boron enoates[J].J Am Chem Soc,1981,103:2127-2129.
[12] Michael A, Walker S. Extending the scope of the Evans symmetric aldol reaction: preparation of anti and “Non-Evans” syn-aldols[J].J Org Chem,1991,56:5737-5750.
[13] Hanson G J, Baran J S, Lindberg T. Stereoselective addition of lithioethyl acetate toN-Boc-l-prolinal.A convenient chiral synthetic building block for the pyrrolizidine alkaloid ring system[J].Tetrahedron Lett,1986,27:3577-3580.
[14] Takayuki S, Kyoko H, Yasumasa H. Stereoselective synthesis of dolastatin 10 and its congeners[J].Tetrahedron,1993,49:1913-1924.
[15] Yasumasa H, Kyoko H, Takayuki S,etal. Efficient stereoselective synthesis of dolastatin 10,an antineoplastic peptide from a sea hare[J].Tetrahedron Lett,1991,32:931-934.
[16] George R P, Douglas D, Burkett M,etal. The dolastatins;18:Stereospecific synthesis of dolaproine 10[J].Synthesis,1993:719-725.
