朊病毒治疗候选药物的研究进展*
2011-11-13钟正伟宋有涛
钟正伟,宋有涛
2.辽宁省动物资源与疫病防治重点实验室,沈阳110036;
3.承德石油高等专科学校化学工程系
疯牛病(BSE)、羊瘙痒症(scrapie)、人库鲁病(kuru)和人克雅氏症(CJD)等都是由朊病毒(prion)引起的一类具有高度传染性和致死性的中枢神经系统疾病[1]。研究表明,细胞内正常朊病毒(PrPC)是一种广泛地表达于脊椎动物细胞表面的内源性糖基化磷脂酰肌醇锚定蛋白,在体内以单体的形式出现,不具有致病性。朊病毒一旦在某种条件下错误折叠后形成致病性朊病毒(PrPSc),其二级结构发生巨大变化,α螺旋降到30%,而β折叠的含量增至45%。同时,致病性朊病毒能以自身为模板,诱导正常朊病毒向致病性构象转化,进一步在分子间通过“交联β折叠片”聚集,形成淀粉状纤维的有毒片断,最终诱导机体内的免疫系统杀死脑神经细胞而导致一系列致死性症状[2-3]。
由于朊病毒的分子致病机理的复杂性,迄今针对疯牛病等朊病毒疾病仍然没有研发出特效的治疗药物和治疗办法,近年来有关抗朊病毒药物的筛选是国际上生物学、医学领域研究的前沿和热点。特别是在美国和欧洲,研究者在动物、细胞和分子水平建立了一系列药物筛选模型来筛选治疗朊病毒疾病的候选药物,并取得了可喜的成果。本文就目前获得的具有抗朊病毒效果的候选药物进行综述,并按照候选药物的结构或功能特点进行了简单的分类,希望能对疯牛病等朊病毒疾病的治疗和药物筛选提供新的思路和方向。
1 多糖类化合物
1.1 糖胺聚糖及其类似物糖胺聚糖是动、植物,特别是高等动物结缔组织中的一类结构多糖,它与核心蛋白共价连接成蛋白聚糖参与细胞粘附、迁移和增殖。糖胺聚糖包括硫酸乙酰肝素、肝素、硫酸皮肤素、硫酸软骨素、硫酸角质素和透明质酸等。其中,硫酸乙酰肝素和肝素具有一定的抗凝血活性,肝素临床上用作抗凝血酶Ⅲ的增强剂。研究表明,硫酸乙酰肝素和肝素在朊病毒感染的小鼠神经母细胞瘤细胞(sc+-MNBs)中可减少PrPSc的聚集[4]。但值得注意的是,硫酸乙酰肝素在无细胞体系中促进PrPC向PrPSc转化,而且转化活性高于硫酸角质素和硫酸软骨素[5]。据此,研究者推测糖胺聚糖可能直接影响PrPSc的形成:在无细胞体系中促进PrPSc的生成,而在细胞环境中由于受到其他因素的影响反而抑制PrPSc的产生。
硫酸乙酰肝素类似物(HM)主要应用于伤口愈合,其化学合成主要是利用不同比例的硫酸基团、羧甲基和苯甲酰胺基团修饰葡聚糖的羟基。细胞实验结果显示,HM2602和HM5004(图1)能够减少朊病毒感染细胞内PrPSc的聚集,而随后的动物实验发现HM2602能够使感染羊瘙痒症仓鼠的发病潜伏期增加14%,并且明显降低羊瘙痒症感染的小鼠脾脏内PrPSc的水平。近期研究发现HM在羊瘙痒症感染粒子(Chandler)感染的鼠神经元细胞(ScGT1-7)中抑制PrPSc聚集的能力与其分子量大小和修饰基团有关,分子量越大,硫酸化程度越高,以及羧甲基比例越低都明显地增加了HM的抗朊病毒效果[6]。虽然分子量增大不利于药物穿透血脑屏障,但通过合理的基团修饰后的HM有可能成为今后有效的抗朊病毒药物之一。
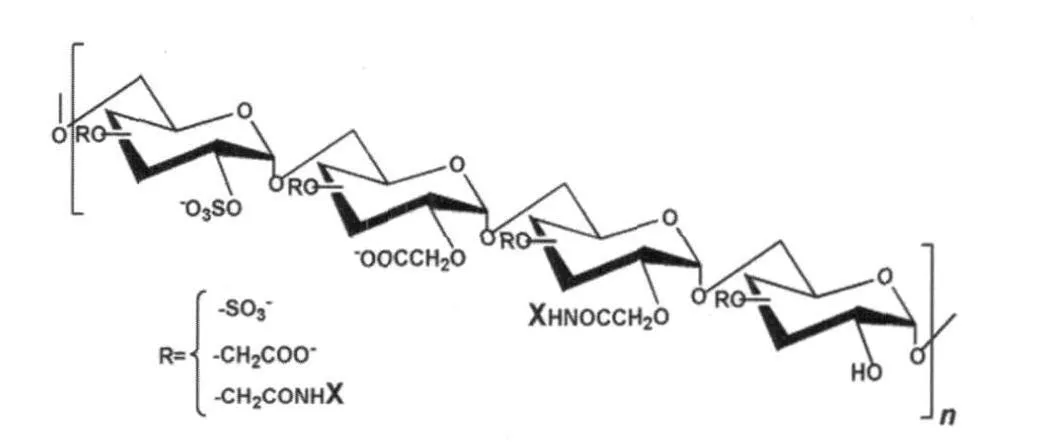
图1 硫酸乙酰肝素类似物的分子结构
X代表苯环,HM2602包含的硫酸基团、羧甲基团和苯甲酰胺基团的比例为0.5∶0.8∶0.2;HM5004包含的硫酸基团、羧甲基团的比例为1.3∶0.5,不含苯甲酰胺基团。
戊聚糖多硫酸酯(PPS)为糖胺聚糖的结构类似物,具有抗凝血与抗炎症活性,临床上用于治疗动物骨关节炎。研究发现PPS与硫酸乙酰肝素的功能相似,在朊病毒感染的细胞内可降低PrPSc的水平,但是在无细胞体系中PPS却加速PrPSc的形成[5]。此外,PPS也延长了羊瘙痒症感染小鼠的发病潜伏期,而且在人克雅氏症感染的鹿细胞(MDB)中抑制PrPSc的聚集[7]。
硫酸葡聚糖 (DS)也是糖胺聚糖的结构类似物,早期研究认为朊病毒发病机制和淋巴网状内皮细胞系统(LPS)有关,而DS可引起LPS的短期破坏,因此DS对嗜淋巴细胞朊病毒株感染的细胞效果尤为明显,经DS处理小鼠脾脏内朊病毒后可明显降低其传染能力;动物实验结果显示分子量为500kDa的DS(DS500)可以延迟腹膜内感染朊病毒小鼠的发病期,也可延迟脑内和腹膜内感染朊病毒仓鼠的发病期。另外,DS500可降低朊病毒感染细胞内PrPSc的水平,但同时也引起未感染朊病毒细胞内正常PrPC的减少[8],因此研究者推测DS500可能通过抑制PrPC的生成从而减少细胞内 PrPSc的水平。
1.2 环糊精 环糊精是一种环状寡糖,一般由6~8个葡糖糖单位通过α-1,4糖苷键连接而成,被作为稳定剂、抗氧化剂、抗光解剂、乳化剂和增溶剂广泛地应用于医药、食品、化妆品等工业中。β-环糊精(图2)具有类似分子伴侣的性质,可以帮助蛋白质折叠,阻止蛋白质的聚集。最近研究发现β-环糊精在羊瘙痒症感染的小鼠N2a22L20细胞中具有清除PrPSc的能力,尤其是β-环糊精被硫酸化修饰后其抗朊病毒活性提高了31倍[9]。
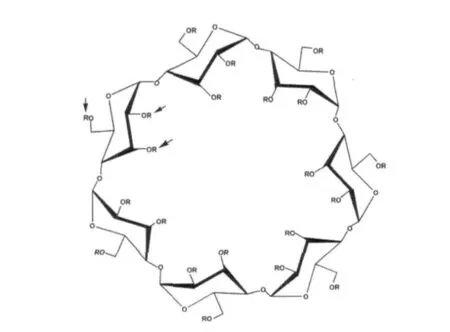
图2 β-环糊精的分子结构箭头所示为硫酸化修饰位置
2 杂环化合物
2.1 卟啉及其金属配合物 卟啉及其金属配合物是一类广泛存在于自然界并具有特殊生理活性的化合物,例如进行生物体内氧气传输的血红素和实现光合作用中能量转移的叶绿素。卟啉临床上主要用于检测和治疗癌症。早期研究发现一些卟啉、酞菁(卟啉的结构类似物)及其金属配合物在羊瘙痒症感染粒子(RML)感染的小鼠神经母细胞瘤细胞(ScN2a)中降低PrPSc的水平,而且在无细胞体系中阻止PrPSc的聚集,其中三种最有效的化合物是四磺酸基酞菁(PcTS)(图 3A)、2,4-二乙二醇基-次卟啉铁(DPG2-Fe3+)(图 3B)和四吡啶基卟啉铁(TMPP-Fe3+)(图3C)。最近有研究表明四苯基卟吩(TPP)、四苯甲酸基卟吩(TCPP)、四苯磺酸基卟吩(TPPS)、四吡啶基卟吩(TMPP)、四苯甲酸基卟啉锰(Mn3+-TCPP)和四吡啶基卟啉锰(Mn3+-TMPP)(图3C)等在两种细胞模型ScN2a与F3(羊瘙痒症感染粒子Fukuoka-1感染的小鼠N2a#58细胞)中抑制PrPSc的聚集,其中作用效果最好的是Mn3+-TMPP(IC50分别为 5nM 与 40nM)[10]。另外,四苯磺酸基卟啉铟(In3+-TSP),在人克雅氏症感染的鹿细胞中可以抑制PrPSc聚集[7],而四(4-N,N,N-三甲基)苯基卟啉铁(Fe3+-TAP)可以4倍地延长腹膜内羊瘙痒症感染粒子(263K)感染的小鼠(tg7)的存活时间[11]。
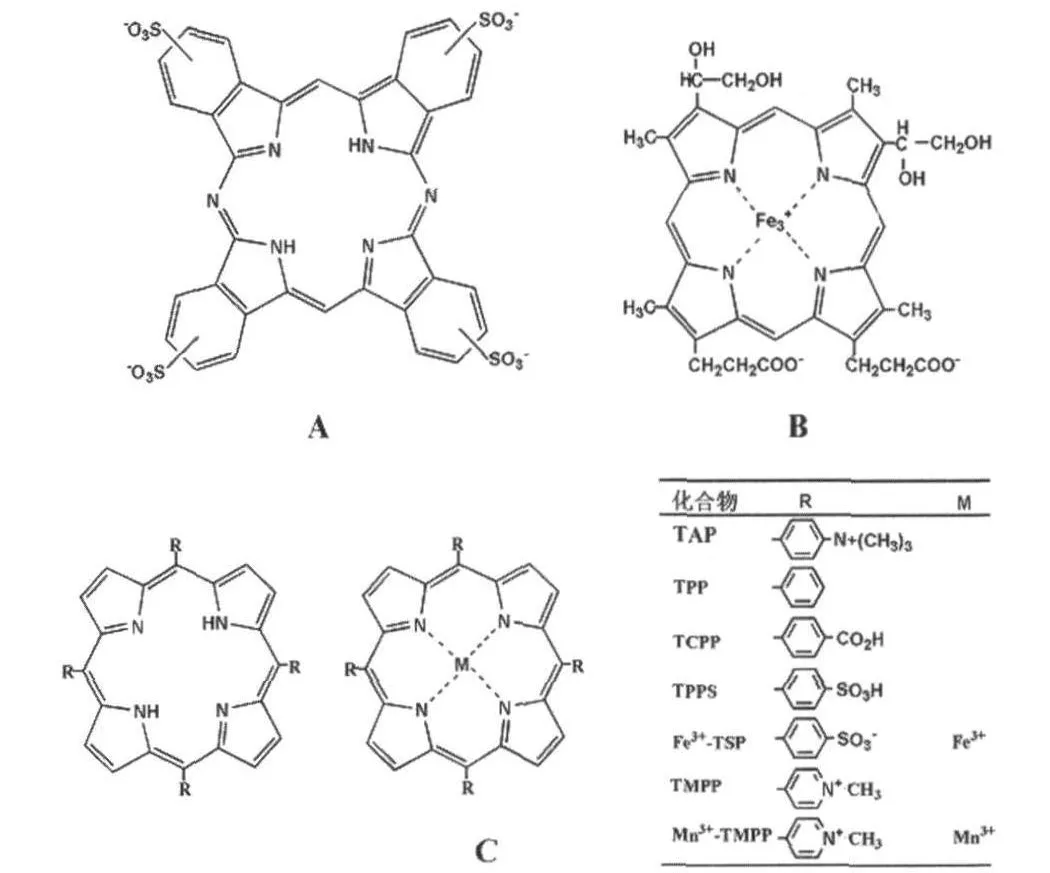
图3 卟啉及其金属配合物的分子结构
2.2 氨噻唑化合物氨噻唑化合物属于甲状腺激素类药物或抗甲状腺药物,近来还被用作腺苷受体拮抗剂。新近研究表明,五种2-氨噻唑化合物(图4)可能通过抑制PrPSc的形成从而降低ScN2a细胞中PrPSc的水平[12]。
2.3 吩噻嗪衍生物和吖啶衍生物 吩噻嗪于1883年合成,曾用作驱虫药,现在其衍生物临床上作为抗精神病药(氯丙嗪)和抗组胺药(异丙嗪)被广泛使用。吖啶衍生物奎纳克林(又称为阿的平)是最早合成的抗疟疾药,由于副作用较大现已停用。2001年Prusiner研究小组发现一系列吖啶和吩噻嗪衍生物在ScN2a细胞中具有不同程度地抑制PrPSc聚集或清除PrPSc的作用,其中最有效的是奎纳克林(IC50为 0.3μ mol/L)和氯丙嗪(IC50为 3μ mol/L)。随后的深入研究表明,吩噻嗪衍生物如氯丙嗪、丙嗪、异丙嗪、硫利达嗪(thioridazine)、三氟拉嗪(trifluoperazine)、丙氯拉嗪(prochlorperazine)和吩噻嗪结构类似物氨砜噻吨(thiothixene)在羊瘙痒症感染粒子(RML)与(22L)感染的小鼠N2a细胞中均可抑制PrPSc的积累。由于吩噻嗪衍生物和吖啶衍生物可作为药物被使用,因而认为其可能是最有潜力的抗朊病毒候选药物,进一步研究发现奎纳克林虽然在无细胞体系和细胞模型中都具有抑制PrPSc聚集的作用,但动物模型研究结果显示奎纳克林没有延长脑和腹膜等部位感染朊病毒小鼠的发病潜伏期。令人惊讶的是,最近的研究结果还显示持续地给患病小鼠注射奎纳克林反而会导致抗药性朊病毒聚集体的产生,这种现象可能有助于解释动物模型中奎纳克林等药物抗朊病毒作用不明显的原因。
2.4 菲啶衍生物 2003年法国Blondel实验室研究了2500多种化合物的抗酵母朊病毒[PSI+]和[URE3]的活性,结果发现菲啶、6-氨基菲啶(6AP)、8-氯-6-氨基菲啶(6A-8CP)、8-三氟甲基-6-氨基菲啶(6A-8tFP)和一类称为kastellpaolitines(KP)(图5)的菲啶衍生物具有很强的抗酵母朊病毒活性,利用ScN2a细胞模型研究发现KP和6AP具有细胞内清除PrPSc的活性[15]。最近的研究表明,抗高血压药胍那苄(GA)(图5)具有抗酵母朊病毒活性,而且还具有在绵羊瘙痒症感染粒子(127S)感染的小鼠MovS6细胞中清除 PrPSc的能力(EC50为7.5μ mol/L),进一步研究GA的卤代物发现其氯代物显著提高了抗酵母朊病毒和细胞内清除PrPSc的活性。此外,动物模型研究显示GA明显延长了127S感染的转基因小鼠(Tg338)的存活时间[16]。随后Blondel等利用亲和吸附法研究发现在酵母中核糖体可能是GA与6AP的作用靶点,GA与6AP不影响核糖体的蛋白合成活性,但可能强烈且专一地抑制核糖体RNA介导的蛋白质折叠活性[15]。
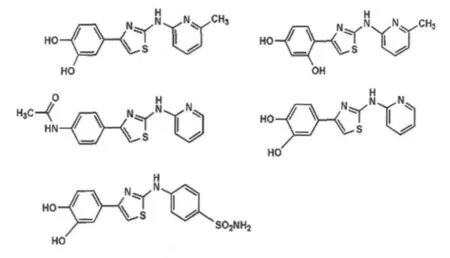
图4 五种2-氨噻唑化合物的分子结构
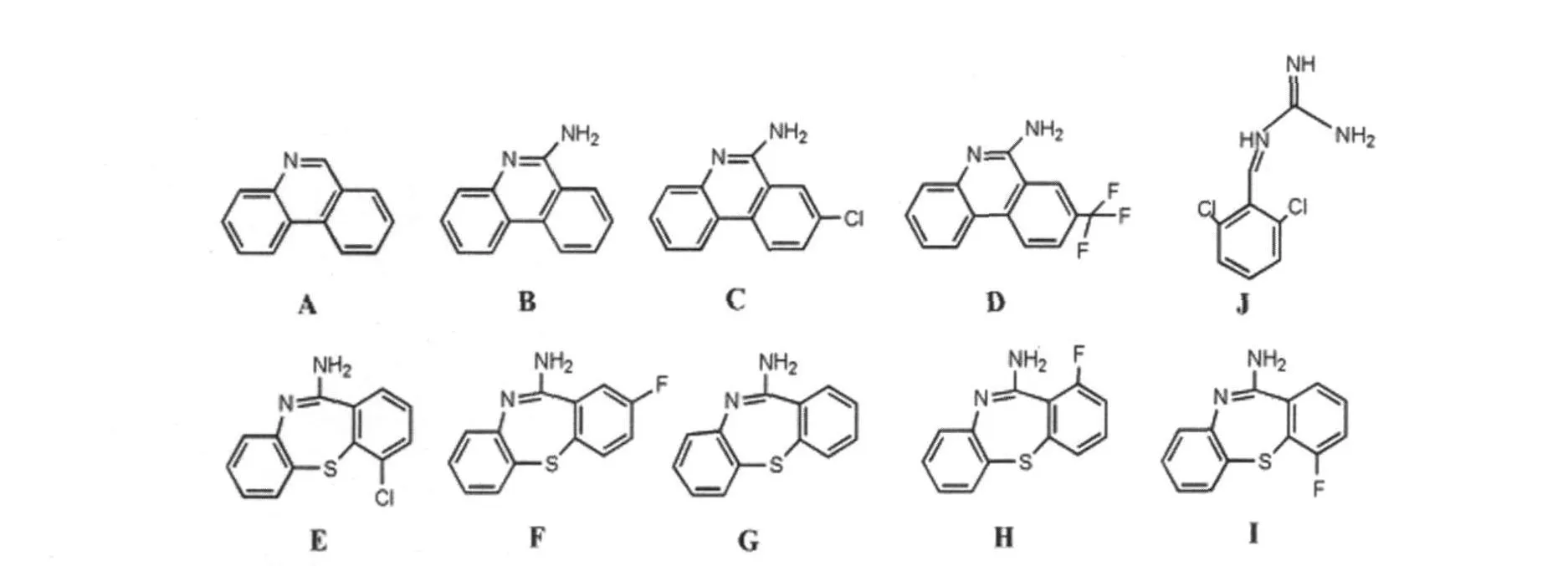
图5 菲啶衍生物和胍那苄的分子结构A :菲啶;B:6-氨基菲啶;C:8-氯-6-氨基菲啶;D:8-三氟甲基-6-氨基菲啶;E:KP1;F :KP2;G:KP3;H:KP4;I:KP5;J:胍那苄。
2.5 生育酚及衍生物 生育酚属于维生素E类,是苯骈二氢吡喃衍生物,具有抗氧化活性,临床上具有抗不育作用。最近,Muyrers等研究发现α-生育酚 、γ-生育酚 、δ-生 育酚和 α-生育酚的三 种衍生物(α-生育酚丁二酸酯、α-生育酚醋酸酯和α-生育酚烟酸酯)在ScN2a细胞中均显示了一定的抗朊病毒作用,其中α-生育酚丁二酸酯的抗朊病毒作用最强(EC50为 7μ mol/L)[17]。
2.6 其他杂环化合物 2005年Bertsch等将重组鼠朊病毒用荧光标记,采用荧光相关光谱(fluorescence correlation spectroscopy,FCS)技术通过96孔板筛选影响 PrPC向 PrPSc转化的药物,在 DI-VERSet1化合物库中发现了80种抑制PrPSc形成的化合物,而且实验证实 6种化合物(293G02、309F02、305E04 、297F03 、313B02 和 260D06)(图 6)在羊瘙痒症感染的N2a细胞中抑制致病性朊病毒的传播,其中四种化合物(293G02、309F02、305E04、297F03)都含有一个共同的核心结构N′-苯亚甲基-苯并酰肼(N′-benzylidene-benzohydrazide)[18]。
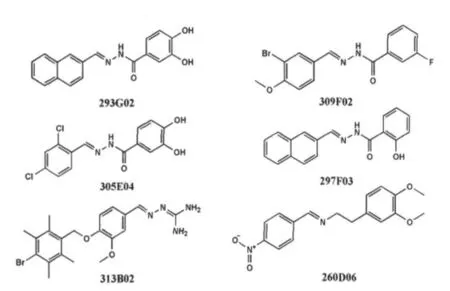
图6 杂环化合物 293G02、309F02、305E04、297F03、313B02和260D06的分子结构
最近,Hosokawa-Muto等利用AutoDock软件设计的虚拟模型在ZINC化合物库中研究了小分子化合物与朊蛋白向致病结构转变过程的相互关系,结果发现205种化合物影响了朊蛋白的构象转变。随后利用Fukuoka-1感染的小鼠GT1-7细胞研究了以上 205种化合物的抗朊病毒活性,实验证实GN8 、GFP23、GFP07、GJP45 和 GJP49(图 7)等 24种杂环化合物具有一定的抑制 PrPSc聚集的作用[19]。
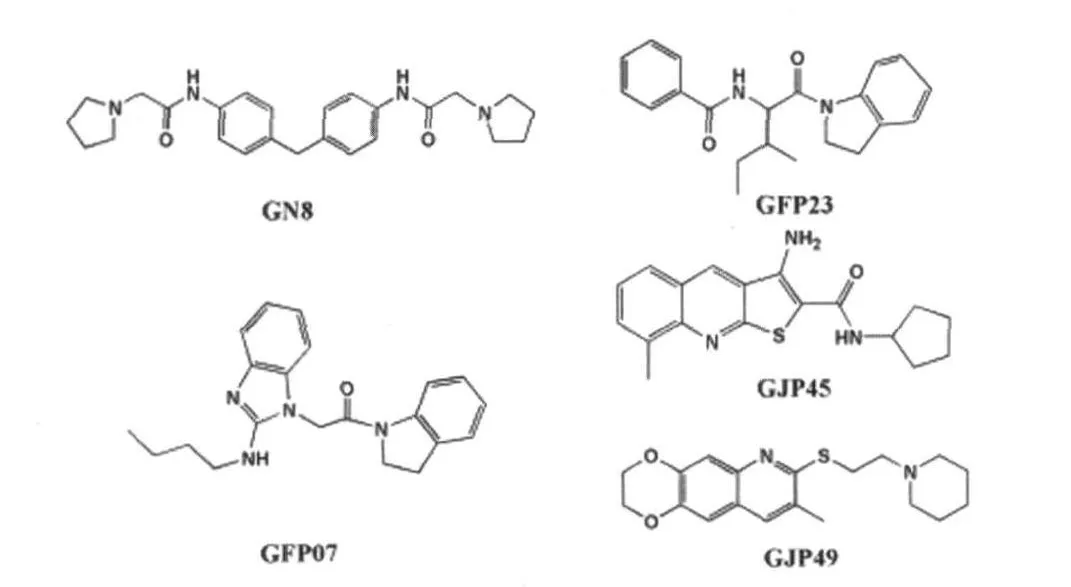
图7 杂环化合物GN8、GFP23、GFP07、GJP45和GJP49的分子结构
刚果红(CR)是医学实验常见的细胞标记染料,广泛地应用于淀粉样脑病的病理学切片染色。CR是目前发现的在多种研究模型(羊瘙痒症感染的细胞模型、无细胞体系和腹腔或脑内感染朊病毒的仓鼠)中具有抗朊病毒效果的化合物。虽然CR存在着细胞毒性和不能通过血脑屏障,限制了其临床用途,但促进了其他淀粉样纤维染料和CR类似物的抗朊病毒作用研究。进一步的研究结果显示,埃文斯红、台盼蓝、天狼星红、硫磺素S和樱草灵等淀粉样纤维染料在无细胞体系中具有很强的抑制PrPSc聚集的效果,但在朊病毒感染细胞内没有抗朊病毒作用。舒拉明(Suramin)结构同源于CR,临床上用于治疗锥虫病。在朊病毒感染的小鼠细胞中发现其通过降低细胞正常PrPC的表达水平从而减少PrPSc的积累;在无细胞体系中舒拉明却导致重组PrP的聚集[20]。
姜黄素是姜黄根的主要成分,结构类似于刚果红,是一种非毒性的抗氧化剂。姜黄素可减少阿尔茨海默症小鼠体内β肽的聚集,在无细胞体系中能抑制PrPSc的形成,并且在RM L感染的N2a细胞中有效地抑制PrPSc的聚集(IC50为10nmol/L),但在22L感染的鼠N2a细胞或绵羊细胞模型中无明显抗朊病毒效果[21]。
阿司咪唑(astemizole)和特非那定(terfenadine)是两种常见的抗组胺药,属于组胺H1受体拮抗剂。研究表明阿司咪唑和特非那定在 RML与22L感染的鼠N2a细胞中均可以抑制PrPSc的积累[13]。遗憾的是,这两种药物穿过血脑屏障的能力不强,从而限制了其应用于治疗传染性海绵样脑病。
3 抗生素类化合物
3.1 两性霉素B(AmB) 两性霉素B是来源于链霉菌属的抗真菌抗生素,其抑菌机理是通过结合真菌细胞膜上的麦角甾醇从而损伤膜的通透性。早期的研究发现AmB(图8)可以延迟人克雅氏症感染的非洲绿猴的发病时间,也可以推迟羊瘙痒症感染的仓鼠与小鼠的发病期,即使在小鼠感染后期,AmB仍然可以延长其存活时间。最近的研究结果显示,AmB的结构类似物16B和3种新的结构类似物(16-19B、8-16A、Tet-aglycone)具有在 22L感染的小鼠N2a22L20细胞中清除PrPSc的能力,其中3种新结构类似物的抗朊病毒活性强于AmB,而16B的抗朊病毒活性与AmB相当,与AmB相比,两性霉素A(AmA)基本不具有抗朊病毒活性[22](图8)。
3.2 四环类抗生素 四环类抗生素是一类广谱抑菌剂,其药理是抑制细菌蛋白质的合成。研究发现,盐酸四环素和盐酸多西环素明显地降低新克雅氏症患者和疯牛病病牛脑匀浆内PrPSc的含量,而且患瘙痒症羊的脑匀浆经盐酸四环素和盐酸多西环素预处理24 h后,可以降低其再感染叙利亚仓鼠的能力。最近研究表明,经皮下注射给药后,盐酸四环素、盐酸多西环素和盐酸米诺环素都可以延长羊瘙痒症感染的叙利亚仓鼠的存活时间;但经脑部注射脂质体包埋的药物后,仅盐酸多西环素和盐酸米诺环素可有效地增加羊瘙痒症感染的仓鼠的半数生存期[23]。
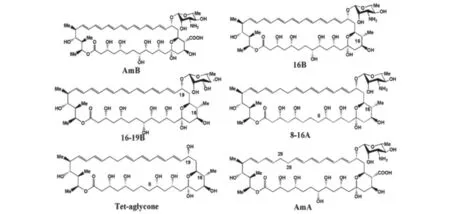
图8 两性霉素B及其结构类似物的分子结构
4 其他化合物
早期研究发现分支聚胺(branched polyamines)在羊瘙痒症感染的ScN2a细胞模型中可以清除PrPSc。遗憾的是,分支聚胺的细胞毒性较强,限制了其临床用途。新近研究显示分支聚乙烯亚胺和分支聚乙二胺经过季铵化作用后,明显降低了其细胞毒性,而且对比未季铵化的分支聚胺,其抗朊病毒效果未发生明显变化[24]。绿茶提取物中的主要多酚类物质表没食子儿茶素没食子酸酯(EGCG)和没食子儿茶素没食子酸酯(GCG)在ScN2a细胞中显著地清除 PrPSc,但强烈地干扰未感染细胞中正常PrPC的表达[25]。研究同时还发现EGCG和GCG将天然伸展的PrPC转变为去污剂不溶的异构体,异构体在细胞内被迅速降解,从而保护细胞不易被PrPSc感染。另外,半胱氨酸蛋白酶抑制剂E-64d能够选择透过细胞膜导致ScN2a细胞中PrPSc降低,而且不影响PrPC的正常代谢[26]。需要注意的是,Fukuuchi等研究卟啉化合物的抗朊病毒作用时发现三种铜螯合剂——新亚铜试剂(Neocuproine)、浴铜灵(Bathocuproine)和联喹啉(2,2′-biquinoline,BQ)在ScN2a与F3细胞中对PrPSc聚集体形成具有抑制作用[10]。最近的研究成果显示 BQ导致ScN2a细胞中PrP的总量减少和抑制PrP的mRNA的表达水平,但不影响未感染的 N2a细胞中PrP的总量[27],因此研究者认为BQ抑制PrPSc聚集体形成的原因是降低了感染细胞中PrP的总量。
5 抗 体
除了常规的疯牛病治疗药物的研发,一个不容忽视的是,近年来对具有抗朊病毒效果的抗体研究发展迅速。2001年Enari等研究发现单克隆抗体6H4(抗体识别表位为鼠PrP第144-152位残基)在感染羊瘙痒症的N2a细胞模型中显著地抑制PrPSc的聚 集(IC50为 2.5 μ g/mL)。2004 年Beringue等利用稳定表达牛PrP的朊病毒感染兔Rov细胞,证实了基因重组小鼠来源的抗人α或β构象 PrP 抗体 ICSM18、ICSM19、ICSM35 、ICSM37和ICSM42在低浓度下能有效地抑制PrPSc的聚集,作用3周后,ICSM35、ICSM37和ICSM42能够500~1 000倍地降低PrPSc的水平,ICSM18和ICSM19能够100倍地降低PrPSc含量,进一步研究证实效果最明显的三种抗体为抗人β构象的PrP抗体,识别表位均为 96-109位残基[28]。最近的研究结果显示,采用人PrP肽段、牛鼠重组PrP和羊瘙痒症纤维(SAF)免疫小鼠所获得的145种单克隆抗体中,有37种抗体能不同程度地降低 N2a细胞(鼠源PrPSc感染的小鼠神经母细胞瘤细胞)和Rov9细胞(羊PrPSc感染的兔上皮细胞)中PrPSc的水平[29]。尽管采用不同免疫原获得的抗体在不同的细胞模型中显示了很强的清除PrPSc的能力,但由于获得的全长抗体分子量较大不利于其穿透血脑屏障因而限制了其在病灶部位发挥作用。因此减小抗体的分子量大小或研究易穿透血脑屏障的抗体越来越受到重视。最近研究发现一种外源表达的单链抗体D18scFv(由抗牛鼠重组PrP抗体D18的VH+VL组成)与ScGT1细胞共培养一周后能降低细胞中PrPSc含量的95%左右[30]。更令人兴奋的是,研究者将D18scFv的cDNA分别克隆在慢病毒(Lentivirus)与腺病毒(AAV2)转导载体上,然后转染ScN2a与ScGT1细胞,并采用免疫印迹技术分别检测两种细胞中PrPSc的含量,结果发现在培养一周后,慢病毒介导的转染体系导致ScN2a与ScGT1细胞中PrPSc的含量分别减少了90%和80%,腺病毒介导的转染体系也导致ScN2a与ScGT1细胞中PrPSc的含量分别减少了20%和5%。虽然这种基于基因治疗的由病毒载体介导的抗体免疫法其安全性还有待进一步研究,但我们有理由相信这可能是今后治疗朊病毒疾病的最佳途径之一。另外,新近研究发现,借助噬菌体展示载体和真核分泌表达系统构建的scFvT2抗体(由抗重组鼠PrP121-231抗体T2的VH+VL组成)表达系统pSecscFvT2,转染ScN2a细胞培养4d后,可明显地降低PrPSc的水平[31]。此外,该研究领域一个突破性的进展是关于来源于骆驼的抗PrP抗体PrioV3的研究:抗体PrioV3在ScN2a细胞可以有效地抑制PrPSc的聚集,它不仅比抗体ICSM35具有更小的细胞毒性,更重要的是,抗体PrioV3可以穿透血脑屏障,其原因可能是骆驼产生的抗体对比常规来源的四链抗体仅由两条重链组成,而且缺少重链的CH1区[32]。
6 结 语
朊病毒治疗候选药物的研究是一个非常活跃的研究领域,研究者们利用无细胞体系、酵母细胞、动物细胞、和模式动物设计了一系列的抗朊病毒药物筛选模型,并利用这些模型筛选获得了大量有潜力的抗朊病毒候选药物(表1)。

表1 目前抗朊病毒候选药物的作用效果和可能机制
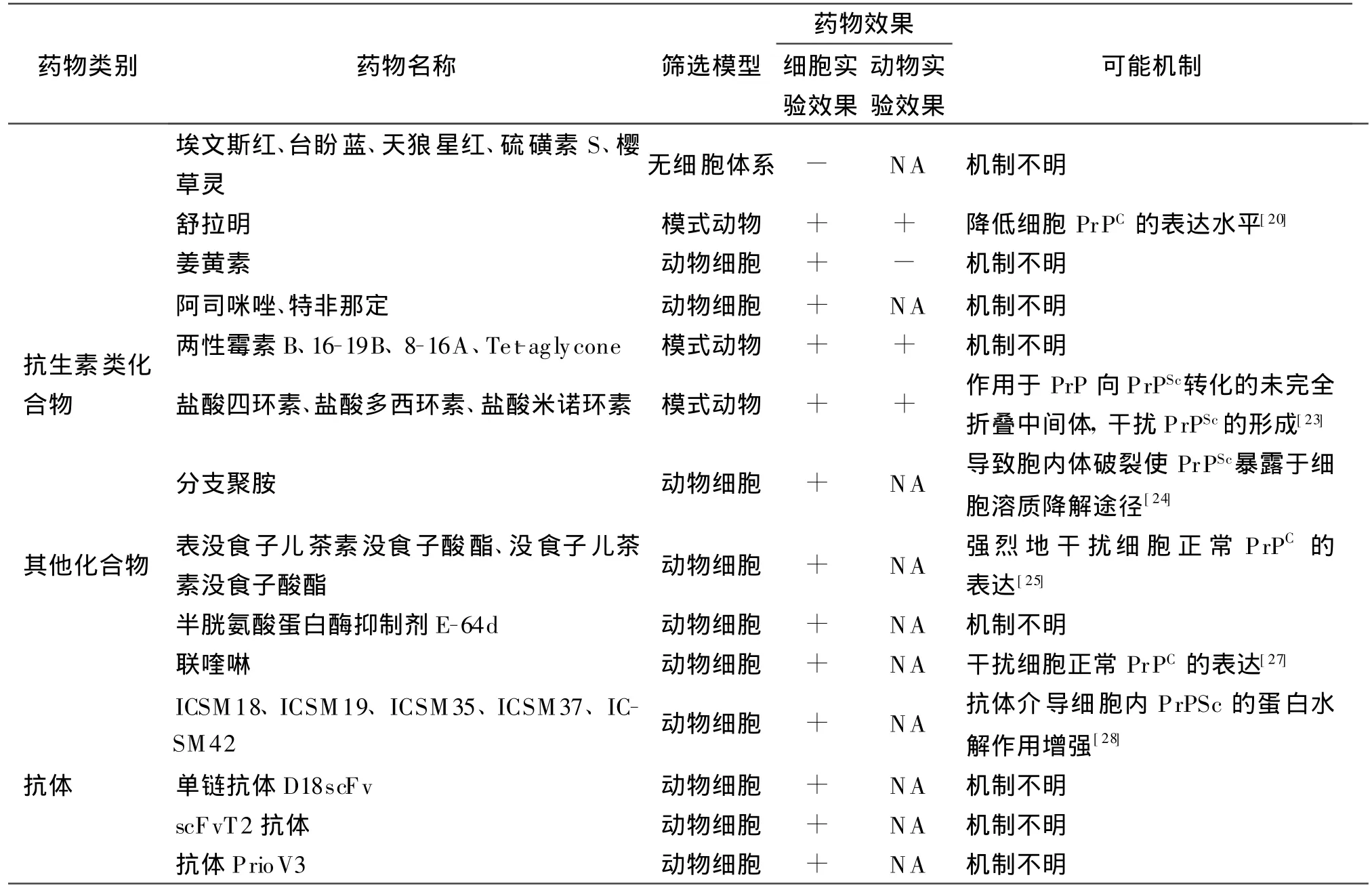
续表
但需要指出的是,许多候选药物在模式动物中的抗朊病毒效果并不十分理想。主要原因是朊病毒疾病主要发生在脑部,而常规候选药物难以穿透血脑屏障发挥作用;其次有些候选药物的细胞毒性较大,限制了其临床用途;再次,不同种属的朊病毒疾病的差异较大,也导致了单一的药物难以发挥广泛地治愈朊病毒疾病的作用。因此,对当前候选药物进行结构修饰,以期提高疗效、降低毒性和增强穿透血脑屏障的能力,可能是今后抗朊病毒药物研究的一个方向。另外,寻找新型抗朊病毒候选药物,以期获得抗朊病毒效果更佳的候选药物也应该成为今后研究的重点,如本实验室借助传统中草药多途径、多方式、多靶点的作用优势,正在进行的从中草药中筛选抗朊病毒候选药物的研究。总之,随着朊病毒疾病发病分子机制研究的进一步深入,抗朊病毒药物研发的进一步发展,相信在不久的将来首个投入临床使用的朊病毒治疗药物即将诞生。
[1]Prusiner S B,Scott M R,Dearmond S J,et al.Prion protein biology[J].Cell,1998,93(3):337-348.
[2]张杰,路伟,刘永生.细胞型朊蛋白(PrPC)研究进展[J].中国人兽共患病学报,2008,24(1):83-86.
[3]Nelson R,Sawaya M R,Balbirnie M,et al.Structure of the cross-beta spine of amy loid-like fibrils[J].Nature,2005,435(7043):773-778.
[4]Caughey B,Brown K,Raymond G J,et al.Binding of the protease-sensitive form of PrP(prion protein)to sulfated glycosaminoglycan and congo red[corrected][J].J Virol,1994,68(4):2135-2141.
[5]Wong C,Xiong L W,Horiuchi M,et al.Sulfated glycans and elevated temperature stimulate PrPSc-dependent cell-free formation of protease-resistant prion protein[J].EMBO J,2001,20(3):377-386.
[6]Ouidja M O,Petit E,Kerros M E,et al.Structure activity studies of heparan mimetic poly anions for anti-prion therapies[J].Biochem Biophys Res Commun,2007,363:95-100.
[7]Raymond G J,Olsen E A,Lee K S,et al.Inhibition of proteaseresistant prion protein formation in a transformed deer cell line infected with chronic wasting disease[J].J Virol,2006,80(2):596-604.
[8]Shy ng S L,Lehmann S,Moulder K L,et al.Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein,PrPC,in cultured cells[J].J Biol Chem,1995,270(50):30221-30229.
[9]Mcevoy K,Mcmahon H E.Antiprion action of new cyclodex trin analogues[J].Biochim Biophys Acta,2009,1790(10):1382-1386.
[10]Fukuuchi T,Doh-Ura K,Yoshihara S,et al.Metal complexes with superoxide dismutase-like activity as candidates for antiprion drug[J].Bioorg Med Chem Lett,2006,16(23):5982-5987.
[11]Kocisko D A,Caughey B.M efloquine,an antimalaria drug with antiprion activity in vitro,lacks activity in vivo[J].J Virol,2006,80(2):1044-1046.
[12]Ghaemmaghami S,May B C,Renslo A R,et al.Discovery of 2-aminothiazoles as potent antiprion compounds[J].J Virol,2010,84(7):3408-3412.
[13]Kocisko D A,Baron G,Rubenstein R,et al.New Inhibitors of Scrapie-Associated Prion Protein Formation in a Library of 2,000 Drugs and Natural Products[J].J Virol,2003,77(19):10288-10294.
[14]Ghaemmaghami S,Ahn M,Lessard P,et al.Continuous quinacrine treatment results in the fo rmation of drug-resistant prions[J].PLoS Pathog,2009,5(11):e1000673.
[15]Tribouillard-Tanvier D,Dos R S,Gug F,et al.Protein folding activity of ribosomal RNA is a selective target of two unrelated antiprion drugs[J].PLoS One,2008,3(5):e2174.
[16]Tribouillard-Tanvier D,Beringue V,Desban N,et al.Antihypertensive drug guanabenz is active in vivo against both yeast and mammalian prions[J].PLoS One,2008,3(4):e1981.
[17]Muyrers J,Klingenstein R,Stitz L,et al.Structure activity relationship of tocopherol derivatives suggesting a novel non-antioxidant mechanism in antiprion potency[J].Neurosci Lett,2010,469:122-126.
[18]Bertsch U,Winklhofer K F,Hirschberger T,et al.Sy stematic identification of antiprion drugs by high-throughput screening based on scanning for intensely fluorescent targets[J].J Virol,2005,79(12):7785-7791.
[19]Hosokawa-Muto J,Kamatari Y O,Nakamura H K,et al.Variety of antiprion compounds discovered through an in silico screen based on cellular-form prion protein structure:Correlation between antiprion activity and binding affinity[J].Antimicrob Agents Chemother,2009,53(2):765-771.
[20]Sim V L,Caughey B.Recent advances in prion chemotherapeutics[J].Infect Disord Drug Targets,2009,9(1):81-91.
[21]Kocisko A D,Engel A L,Harbuck K,et al.Comparison of protease-resistant prion protein inhibitors in cell cultures infected with two strains of mouse and sheep scrapie[J].Neurosci Lett,2005,355:106-111.
[22]Soler L,Caffrey P,Mcmahon H E.Effects of new amphotericin analogues on the scrapie isoform of the prion protein[J].Biochim Biophys Acta,2008,1780(10):1162-1167.
[23]Luigi A D,Colombo L,Diomede L,et al.The efficacy of tetracyclines in peripheral and intracerebral prion infection[J].PLoS One,2008,3(3):e1888.
[24]Lim Y B,May s C E,Kim Y,et al.The inhibition of prions through blocking prion conversion by permanently charged branched polyamines of low cytotoxicity[J].Biomaterials,2010,31(8):2025-2033.
[25]Rambold A S,Miesbauer M,Olschew ski D,et al.green tea eatract interfere with the stress-protective activity of PrPC and the formation ofPrPSc[J].J Neurochem,2008,107:218-229.
[26]Doh-Ura K,Iwaki T,Caughey B.Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation[J].J Virol,2000,74(10):4894-4897.
[27]Fukuuchi T,Okuda K,Yoshihara S,et al.A Candidate Anti-Prion Disease Agent,2,2-Biquinoline Decreases Expression of Prion P rotein and mRNA in Prion-Infected Cells[J].J Health Sci,2009,55(4):586-592.
[28]Beringue V,Vilette D,Mallinson G,et al.P rPSc binding antibodies are potent inhibitors of prion replication in cell lines[J].J Biol Chem,2004,279(38):39671-39676.
[29]Feraudet C,M orel N,Simon S,et al.Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells[J].J Biol Chem,2005,280(12):11247-11258.
[30]Campana V,Zentilin L,Mirabile I,et al.Development of antibody fragments for immunotherapy of prion diseases[J].Biochem J,2009,418(3):507-515.
[31]Shimizu Y,Kaku-Ushiki Y,Iwamaru Y,et al.A novel antiprion protein monoclonal antibody and its single-chain fragment variable derivative with ability to inhibit abnormal prion protein accumulation in cultured cells[J].Microbiol Immunol,2010,54:112-121.
[32]Jones D R,T aylo r W R,Bate C,et al.A Camelid Anti-PrP Antibody Abrogates PrPSc Replication in Prion-Permissive Neuroblastoma Cell Lines[J].P LoS One,2009,5(3):e9804.
