23株HBeAg阳性慢性乙型肝炎患者病毒的全基因组分析*
2011-08-21李志军苏智军林成祖郭如意林万松
李志军,苏智军,林成祖,郭如意,林万松
2.福建医科大学附属泉州第一医院感染病科,泉州362000;
3.福建医科大学肿瘤微生物学福建省重点实验室,福州350001
23株HBeAg阳性慢性乙型肝炎患者病毒的全基因组分析*
李志军1,苏智军2,林成祖2,郭如意2,林万松3
目的研究HBeAg阳性慢性乙型肝炎患者HBV变异特点。方法 PCR扩增并克隆HBeAg阳性慢性乙型肝炎患者血清中HBV全基因组DNA,测序并进行基因结构分析。结果 获得23株HBV全基因组DNA,它们均属于C或B基因型。与中国HBV B、C基因型参照序列相比,HBeAg阳性慢性乙型肝炎患者来源的HBV在表面抗原、P蛋白、X蛋白的反式激活区及增强子II/核心启动子区发生了一些有意义的共有变异。结论 HBV变异可能与HBeAg阳性慢性乙型肝炎的发生、发展有关。
乙型肝炎病毒;基因变异;全基因组
2.福建医科大学附属泉州第一医院感染病科,泉州362000;
3.福建医科大学肿瘤微生物学福建省重点实验室,福州350001
据WHO估计,全球大约有20亿人感染了乙型肝炎病毒(HBV),约有3.5亿人患有慢性感染[1]。我国是乙肝大国,HBV感染率为50.1%,乙肝表面抗原携带率约占总人口的7.18%,以此计算,全国约有9300万人携带 HBV,其中乙肝患者大约有3000万[2]。研究显示,HBV一些特定位点的变异可以引起慢性乙型肝炎抗病毒治疗的失败。另外,由于大多数的研究是以HBV的部分片段或特定位点作为研究对象,并不能反映病人体内病毒的真实状态,具有一定的局限性。为了更好的理解HBV的生物学特征,本文采用全基因克隆的方法,了解HBeAg阳性慢性乙型肝炎患者HBV变异特点。
1 材料与方法
1.1 实验材料
1.1.1 血清 标本来自福建医科大学附属泉州第一医院11例和福州市传染病院12例HBeAg阳性慢性乙型肝炎患者2006-2009年血清共23例。所有病例诊断按2000年中华医学会传染病与寄生虫病学分会、肝病学分会西安会议修订的病毒性肝炎防治方案诊断标准。
1.1.2 主要试剂 pSure-T载体购自北京博迈德科技发展有限公司,DH5α菌株购自Invitrogen公司,蛋白酶 K、酵母tRNA购自Sigma公司,限制性内切酶HindⅢ、T4 DNA ligase购自Biolab公司。Platinum Taq DNA Polymerase High Fidelity购自Ivitrogen公司,胶回收及质粒小量抽提试剂盒购自上海超世生物科技有限公司。
1.2 方法
1.2.1 血清 HBV DNA 抽提 100 μ L血清加入300 μ L TES 液 (10 mmol/L Tris-HCl,pH8.0,5 mmol/L EDTA,0.5%SDS,50μ g Protease K),混匀,65℃消化过夜,等体积苯酚/氯仿及氯仿抽提各1次,上清加入2倍体积无水乙醇、1/10体积3 mol/L醋酸钠(pH 4.8)及20μ g酵母tRNA,DNA沉淀以70%乙醇漂洗1次并溶于50μ L灭菌双蒸水中。
1.2.2 HBV DNA全基因扩增 引物参照Günther等方法[3]设计并改进,正向引物序列:5′CCCAAGCT TGAGCTCT TCT TT TTCACCTC TGCCTAATCA 3′, 反 向 引 物 序 列:5′CCCAAGCT TGAGCTCT TCAAAAAGTTGC ATGGTGCTGG 3′。PCR扩增条件:反应体积25μ L。各成分的终浓度为:2.5 mmol/L MgSO4,0.2 mmol/L dNTP,正反向引物各20pmol,lxPCR缓冲液,5 uL抽提HBV DNA,1U高保真DNA聚合酶。扩增采用热启动方式,即:反应先经预变性95℃3min,待温度上升至80℃后加入T aq/pwo聚合酶,随后进入循环。PCR扩增程序为:95℃40s,61℃1min,68℃3min(第 1-20循环);94℃40s,61℃1min,68℃3min(第21-35循环,引物延伸时间每循环递增5s);最后68℃延伸10min。
1.2.3 PCR产物克隆及重组子的筛选 PCR产物回收后与pUC18在4℃连接16 h,转化感受态大肠杆菌DH5α。在含氨苄青霉素及X-gal、IPTG平板上挑取白色菌落,碱裂解法抽提DNA,以HindⅢ酶切鉴定。
1.2.4 HBV DNA基因组序列测定 以ABI371型DNA自动荧光序列分析仪进行序列测定。所用测序引物包括pUC18通用引物及HBV DNA特异性引物。
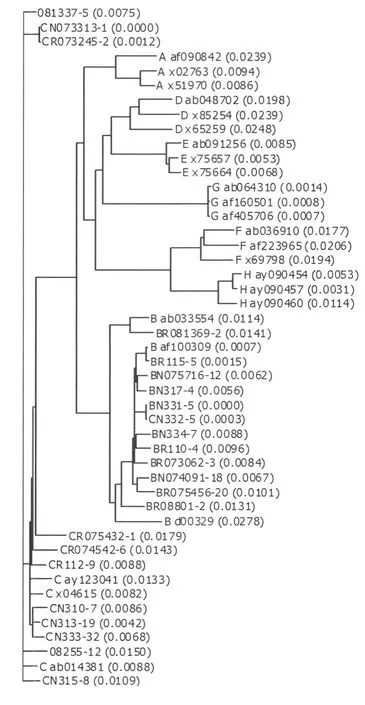
1.2.5 HBV DNA及各编码区氨基酸分析 基因型分析:采用Vector NTI 10.0分析软件对所分离的HBV DNA通过序列比较进行基因型归类,基因型分析采用的标准株为:基因型 A(X02763、X51970、AF090842),基 因 型 B (D00329、AF100309、AB033554),基因型 C(M12906、X04615、AB014381),基 因 型 D (X65259、AB048702、X85254),基因型 E(X75664、X75657、AB091256),基因型 F(AB036910、AF223965、X69789),基因型 G(AF160501、AB064310和AF223965),基因型 H(AY090454、AY090457 和AY090460)。实验结果以中国HBV B、C基因型参照序列[4]为标准序列。
2 结 果
2.1 乙型肝炎病毒全基因组DNA扩增及克隆从23份HBeAg阳性的慢性乙型肝炎患者血清中扩增出3 215 bp的HBV全基因组。将HBV全基因组DNA通过T-A克隆至pSure-T载体,以ABI 3730XL全自动荧光序列分析仪进行序列测定。
2.2 基因型 以已知的标准A-H型HBV全基因组DNA为参照,以VectorNTI10.0软件对所获得的全基因组 HBV DNA进行基因型分析,结果显示,在所获得的23株HBV中,B基因型为12例,C基因型为11例。
2.3 乙型肝炎病毒各编码区氨基酸及基因调控区变异 本研究的23例(B12,C11)血清标本中,与中国HBV B、C基因型参照序列相比,B基因型的主要变异位点分别位于:nt753T,nt1368A,nt1752G,nt2699G,nt2712T,nt3097C;C基因型的主要变异位点分别位于:nt31C,nt934C,nt1762T,nt1764A,nt2681G,nt2699G,nt3026C,nt3166C。
2.3.1 preS及S区氨基酸变异在preS1区,B基因型的HBV株在60位氨基酸发生A→V突变,C基因型的HBV株在84位氨基酸发生I→L突变见表1,在preS2区,虽然有核苷酸的变异,但却没有氨基酸突变。另外,HBV B基因型在S区第200位氨基酸发生F→Y变异。
2.3.2 P区氨基酸变异HBV B基因型在P区第136位氨基酸发生Y→H突变,此突变位于 TP区。C基因型在P区264位氨基酸发生P→H突变,此突变位于space区;在314位氨基酸发生P→S突变,位于逆转录酶(RT)区;另外,在615位氨基酸发生 L→I突变,位于RH 区。
2.3.3 增强子II/核心启动子区变异 增强子II(1685-1773nt)和核心启动子区(1742-1849nt)重叠区域有3个位点的氨基酸变异。分别为B基因型1752位核苷酸出现G→A变异,C基因型1762位核苷酸及1764位核苷酸分别出现出现T→A及A→G变异。
2.3.4 X区变异 X蛋白的编码区(1374-1838nt)与增强子II/核心启动子区交叉重叠。增强子II/核心启动子区在1752、1762、1764位核苷酸的变异导致X蛋白在116、130、131位氨基酸发生变异。上述130、131位氨基酸发生变异位于X蛋白的反式激活区内(120-140aa)。
3 讨 论
慢性乙型肝炎的发生过程是HBV与宿主细胞相互作用与共同演变的过程,HBV的生物学特性可显著影响其致病过程。
慢性乙型肝炎病毒变异在不同地区存在很大差异,既有基因型间的差异,也有个别核苷酸位点的差异,我国流行株主要为B、C基因型。本研究获得的23株HBV均为B或C基因型,与中国大陆慢性乙型肝炎的HBV基因型一致。HBV基因型与慢性乙型肝炎的关系有待进一步研究。
preS1区,B基因型60位氨基酸A→V突变位点、C基因型84位氨基酸I→突变位点为preS1抗体的结合区域(58-100aa)[5],该区域氨基酸变异可能影响病毒清除,与病毒的持续感染有关。研究结果显示包膜蛋白变异位于已知的抗体结合区域或B/Th/Tc细胞表位内,提示HBV的免疫逃逸可能是乙型肝炎慢性化的原因之一。由于TP区与干扰素疗效密切相关,因此P基因TP区多个位点的变异对干扰素治疗的影响有待进一步研究;同样位于RT、RH区的变异对慢性乙型肝炎的影响需要进一步加以研究。
1762位点A-T和1764位点G-A双突变(A1762T,G1764A)为核心启动子最常见的变异。核心启动子区突变也能阻止HBeAg的产生,而不会影响HBV的复制或核心抗原的表达,只是选择性下调前核心mRNA的转录,但不影响前基因组RNA[6]。且与前核心区变异不同,检测发现核心启动子变异在HBeAg阴性和HBeAg阳性患者中的比例类似[7]。本研究中,这些位点却并没有发生上述所说的变异,可能与所选病例都为 HBeAg阳性患者有关。
X蛋白可反式激活包括原癌基因在内的多种细胞及病毒基因[8],与肝癌的发生有关。由于X蛋白编码区与增强子II及核心启动子相重叠,增强子II及核心启动子的核苷酸变异可引起相应位置X蛋白氨基酸的变异,本研究中130、131位氨基酸位于X蛋白的反式激活区,这些氨基酸变异与X蛋白的激活活性有待进一步研究。
[1]Ganem D,Prince A M.Hepatitis B virus infection-Natural history and clinical consequences[J].N Engl J M ed,2004,350:1118-1129.
[2]Liang X,Bi S,Yang W,et al.Epidemiological serosurvey of Hepatitis B in China-Declining HBV prevalence due to Hepatitis B vaccination[J].Vaccine,2009,27(47):6550-6657.
[3]Gunther S,Li BC,Miska S,et al.A noval method for efficient amplification of the whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutations in immunosuppressed patients[J].J Virol,1995,69:5437-5444.
[4]吴光华,丁惠国,曾长青.公共核酸数据库乙肝病毒全基因组序列概况和中国HBV参照序列的建立[J].自然科学进展,2008,18(2),121-129.
[5]Fiordalisi G,Ghiotto F,Castelnuovo F,et al.Analy sis of the hepatitis B virus genome and immune response in HBsAg,anti-HBs positive chronic hepatitis[J].J Hepat,1994,20:487-493.
[6]Buckwold VE,Xu Z,Chen M,et al.Effects of a naturally occurring mutation in the hepatitis B virus basal co re promoter on precore gene expression and viral replication[J].Virol,1996,70:5845-5851.
[7]Wang Y,Wei L,Jiang D,et al.In vitro resistance to interferonalpha of hepatitis B virus with basic core promoter double mutation[J].Antiviral Research,2007,75:139-145.
[8]Heermann K H,Goldmann U,Schwartz W,et al.Large surface proteins of hepatitis B virus containing the pre-s sequence[J].J Virol,1984,52(2):396-402.
Structural analysis of 23 full-length hepatitis B virus genomes isolated from HBeAg positive chronic hepatitis B patients
LI Zhi-jun,SU Zhi-jun,LIN Cheng-zu,GUO Ru-yi,LIN Wan-song
(Department of Infectious Diseases,Quanzhou First Hospital Af filiated
toFujian Medical University,Quanzhou362000,China)
To study the characteristics of full-length Hepatitis B Virus(HBV)genomes isolated from HBeAg positive chronic hepatitis B(CHB)patients.Amplified the full-length HBV genomes by PCR from the serum of HBeAg positive CHB patients,sequenced and analyzed the structure of HBV genomes.Obtained twenty-three full-length HBV DNAs from the different CHB patients.Phylogenetic analysis revealed that all HBV strains could be categorized into genotype B or C.Structural analysis showed that,as compared to standard strains of Chinese HBV B、C subtypes,HBV obtained shared meaningful consensus mutations in B/T cell epitopes of surface and P protein,transactive domain of X protein and enhancer II/core promoter regions.Genotype and gene mutation of HBV may closely correlated with the carcinogenesis of HBV-related chronic hepatitis B.
Hepatitis B virus;full-length Gene mutation genomes
R373
A
1002-2694(2011)08-0728-03
*国家自然科学基金资助课题(30840069);2008年福建医科大学科研项目资助课题(FZS08002)和2007年泉州市科技计划项目资助课题(2007Z15)联合资助
苏智军,Emial:su2366@sina.com
1.福州市第一医院,福州 350001;
2011-02-17;
2011-06-12
