微乳液相色谱法同时测定大黄中5种蒽醌类衍生物的含量Δ
2011-08-06何素珍张忠义张守尧南方医科大学珠江医院药剂科广州市510282
何素珍,张忠义,张守尧(南方医科大学珠江医院药剂科,广州市510282)
大黄为蓼科植物掌叶大黄Rheum palmatumL.、唐古特大黄R.tanguticumMaxim.ex Balf.或药用大黄R.officinaleBaill.的干燥根及根茎,具有泻下、通瘀导滞、抗菌消炎、止痛止血、抗肿瘤、抗氧化和降血脂等作用[1],并应用于临床多种危重急症的抢救治疗[2]。大黄的主要有效成分为蒽醌类衍生物,包括芦荟大黄素、大黄酸、大黄素、大黄酚和大黄素甲醚以及与葡萄糖结合成的苷[3]。微乳液相色谱(MELC)法是一项液相色谱新技术[4],它不仅存在常规色谱的分离机制,还存在微乳液滴内外相之间的分配[5]。其与常规高效液相色谱(HPLC)法相比,具有分离效率高和分离速度快的优越性,可用于复杂样品的分离;血清、尿液经过简单的稀释后便可以直接进样分析,无需预处理;在梯度洗脱和低紫外波长(190 nm)检测方面也有一定的优势[6~10]。对于MELC法的应用,主要涉及化学药品中多种成分、药物制剂以及生物样品的分析[11,12],而用于中药成分的分析较少。笔者采用MELC法同时测定了大黄中5种蒽醌类衍生物的含量,方法简便、准确、可靠,取得了满意结果。
1 仪器与试药
Agilent 1100型HPLC仪,配二极管阵列检测器及化学工作站(美国安捷伦科技有限公司);KQ-600DE型数控超声仪(昆山市超声仪器有限公司,功率:600 W,工作频率:40 kHz)。
芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚对照品和掌叶大黄、唐古特大黄药材(中国药品生物制品检定所,批号 分 别 为 110795-200504、110757-200206、110756-200110、110796-200513、 110758-200610、 121249-200402、 120902-200609);药用大黄(购于广州市药材公司,经南方医科大学珠江医院药剂科周本杰主任药师鉴定为真品);十二烷基硫酸钠(SDS,化学纯,西陇化工股份有限公司,批号:0911191);甲醇、乙腈(色谱纯,上海凌峰化学试剂有限公司);水为注射用水,其余试剂均为分析纯。
2 方法与结果
2.1 溶液的制备
2.1.1 混合对照品溶液 取芦荟大黄素、大黄酸、大黄素、大黄酚和大黄素甲醚对照品各适量,精密称定,置于50 mL容量瓶中,加适量甲醇超声溶解,再加甲醇至刻度,摇匀,即得混合对照品溶液。临用前用0.45 μm滤膜过滤,取续滤液进样。
2.1.2 供试品溶液 取干燥后的各大黄药材粉末(过四号筛)约0.3 g,精密称定,置具塞烧瓶中,精密加入甲醇25 mL,称定重量,加热回流1 h,放冷,再称定重量,用甲醇补足失重,摇匀,滤过。精密量取续滤液5 mL,置烧瓶中,挥去溶剂,加8%盐酸溶液10 mL,超声使溶解,再加氯仿10 mL,加热回流1 h,放冷,转移至分液漏斗中,用少量氯仿分次洗涤烧瓶,洗液并入分液漏斗中,分取氯仿层,酸液再用氯仿提取3次,每次10 mL,合并氯仿液,挥去溶剂,残渣加少量甲醇使溶解,定量转移至10 mL容量瓶中,加甲醇至刻度,摇匀,即得供试品溶液。临用前用0.45 μm滤膜过滤,取续滤液进样。
2.2 微乳流动相的制备
将SDS、正丁醇、正辛烷及含0.5%三乙胺的水溶液按顺序混合,超声15 min,即得透明稳定的微乳,用磷酸调pH值至3.0,经0.45 μm滤膜过滤,备用。
2.3 色谱条件
色谱柱:Hypersil ODS2(250 mm×4.6 mm,5 μm);流速:1 mL·min-1;柱温:28 ℃;检测波长:254 nm;流动相:2.5%(W/V)SDS-0.1%(V/V)正辛烷-8.0%(V/V)正丁醇-0.5%(V/V)三乙胺(磷酸调pH值至3.0);进样量:15 μL。色谱见图1。
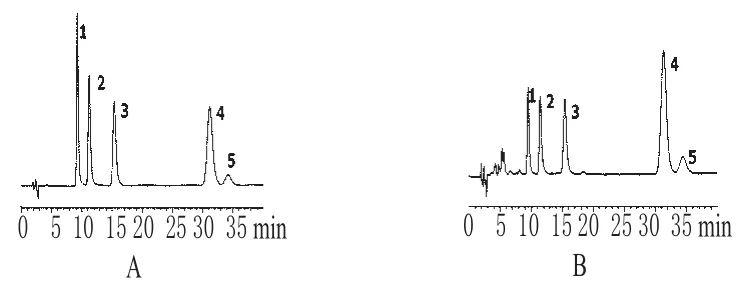
图1 微乳液相色谱图Fig 1 Microemulsion liquid chromatograms
2.4 线性关系考察
精密量取“2.1.1”项下混合对照品溶液0.5、1.0、2.0、3.0、4.0、5.0 mL,置6个10 mL容量瓶中,加甲醇稀释至刻度,摇匀,按“2.3”项下色谱条件,取15 μL进样测定,每个混合对照品溶液进样3次。分别以浓度(X,μg·mL-1)为横坐标,对应的平均峰面积积分值(Y)为纵坐标,进行线性回归,得出大黄中5种蒽醌类衍生物的回归方程,详见表1。

表1 5种蒽醌类衍生物对照品的标准曲线Tab 1 Standard curves of 5 anthraquinone derivatives
2.5 精密度试验
取“2.4”项下芦荟大黄素浓度分别为18.8、37.6 μg·mL-1的混合对照品溶液适量,连续进样6次。结果,各化合物峰面积的RSD(n=6)分别为芦荟大黄素0.49%、0.91%,大黄酸0.48%、0.72%,大黄素0.69%、0.78%,大黄酚0.86%、0.94%,大黄素甲醚1.29%、1.22%,表明仪器精密度良好。
2.6 稳定性试验
取“2.4”项下芦荟大黄素浓度为28.2 μg·mL-1的混合对照品溶液适量,室温避光,每隔1 h进样一次,重复7次。结果,各化合物峰面积的RSD(n=7)分别为芦荟大黄素0.28%、大黄酸0.57%、大黄素0.58%、大黄酚0.38%、大黄素甲醚1.23%,表明大黄中5种蒽醌类衍生物在7 h内稳定。
2.7 加样回收率试验
取已知含量的样品适量,共6份,精密称定,精密加入一定量的混合对照品溶液,按“2.1.2”项下方法制成供试品溶液,按“2.3”项下色谱条件进样测定,计算加样回收率,结果见表2。
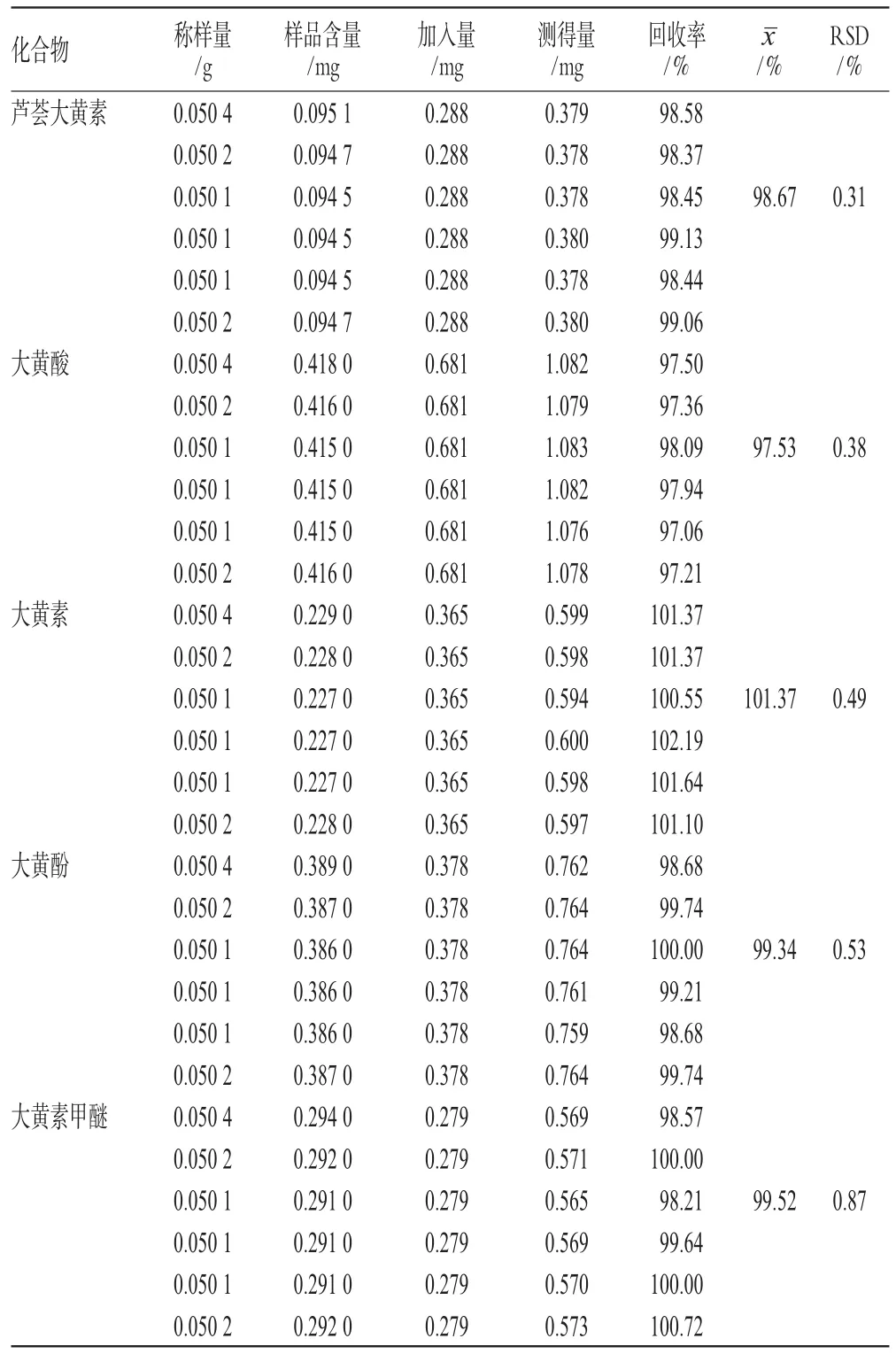
表2 5种蒽醌类衍生物的加样回收率试验结果(n=6)Tab 2Results of recovery tests for 5 anthraquinone(n=6)
2.8 重复性试验
取同一批大黄药材粉末(过四号筛)6份,各约0.3 g,精密称定,按“2.1.2”项下方法制备供试品溶液,按“2.3”项下色谱条件进样测定。结果,各化合物平均含量的RSD(n=6)分别为芦荟大黄素0.38%、大黄酸0.52%、大黄素0.85%、大黄酚1.03%、大黄素甲醚0.48%,表明本方法重复性良好。
2.9 样品含量测定
取3种大黄药材粉末(过四号筛)各约0.3 g,精密称定,按“2.1.2”项下方法制备供试品溶液,按“2.3”项下色谱条件进样测定,计算大黄中5种蒽醌类衍生物的含量,结果见表3。
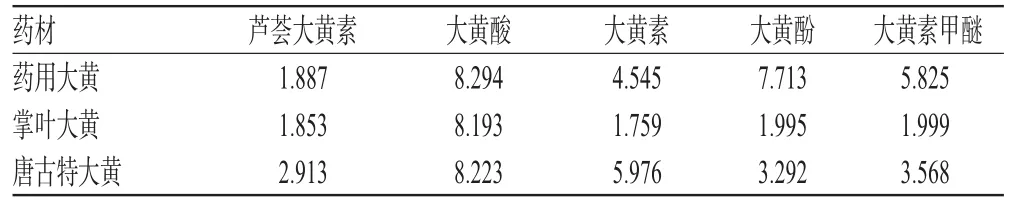
表3 MELC法测定3种大黄中5种蒽醌类衍生物的含量结果(mg·g-1)Tab 3 Results of content determination of 5 anthraquinone in 3 kinds of Rhei Radix Et Rhizoma by MELC(mg·g-1)
3 讨论
3.1 表面活性剂浓度对分离的影响
在微乳稳定区域范围内,考察SDS浓度在20.0~35.0 g·L-1范围变化时的试验结果,发现随着SDS浓度增加,芦荟大黄素与大黄酸的分离度降低,保留时间相应缩短;当SDS浓度在25.0 g·L-1时,分离效果和保留时间最为理想,故确定表面活性剂SDS的试验浓度为25.0 g·L-1。
3.2 助表面活性剂含量对分离的影响
改变助表面活性剂正丁醇的含量,对保留时间和分离效果有影响。笔者考察了正丁醇含量在4.0%~15.0%范围变化时的试验结果,发现随着正丁醇含量的增加,保留时间相应缩短。综合考虑组分分离度和保留时间,确定助表面活性剂正丁醇的浓度为8.0%。
3.3 油相含量对分离的影响
改变油相正辛烷的含量,可以改变物质的保留时间和分离度。笔者考察了正辛烷含量在0.1%~1.4%范围变化时对分离效果的影响,发现随着正辛烷含量的降低,芦荟大黄素与大黄酸、大黄酚与大黄素甲醚的分离度相应提高,保留时间相应延长;当正辛烷的含量为0.1%时,分离效果最理想,故确定油相正辛烷的含量为0.1%。
3.4 pH值对分离的影响
在微乳体系中,用磷酸调流动相pH值在2.5~7.0范围,考察试验结果,发现改变pH值主要是影响芦荟大黄素的保留时间,随着pH值的增加,芦荟大黄素的保留时间相应缩短;当pH值为3.0时,效果最为理想,故选择流动相pH值为3.0。
3.5 添加剂对分离的影响
采用SDS-正辛烷-正丁醇-0.5%三乙胺微乳流动相,分别添加有机溶剂,使成微乳-甲醇(95∶5,V/V)、微乳-乙腈(95∶5,V/V)流动相进行试验。结果显示,加入有机添加剂后,对组分分离度无影响,但保留时间延长。
本试验建立了同时测定大黄药材中5种蒽醌类衍生物含量的微乳液相色谱法。结果表明,5种蒽醌类衍生物达到基线分离,精密度在0.48%~1.29%之间,准确度高;加样回收率在97.53%~101.37%之间;具有较宽线性范围和良好的线性关系(r在0.998 7~0.999 9之间)。同时也表明,采用微乳流动相同样可以分离成分复杂、性质相近的中药里的多种成分,而且有机溶剂用量少、毒性低、相对的费用和污染较少,可成为中药质量控制方法的新选择。
[1] 李 娟,李 坚.大黄药理作用研究及临床应用概况[J].实用医药杂志,2006,23(9):1 132.
[2] 涂久生,于立军.大黄在危重急症临床应用中的最新进展[J].中国药房,2008,19(36):2 868.
[3] 吕慧英,赵晨曦,梁逸曾,等.5种大黄蒽醌类衍生物的同时测定及应用[J].时珍国医国药,2009,20(10):2 490.
[4] 李 宁,黄光亮,李玉兰,等.微乳液相色谱法同时测定山楂叶提取物中4种黄酮成分[J].分析化学研究简报,2009,37(12):1 791.
[5] 张守尧,周苏秦,王秉钧,等.微乳液相色谱法测定虎杖中2种成分的含量[J].今日药学,2009,19(2):41.
[6] Marsh A,Clark B,Altria K.A review of the background,operating parameters and applications of microemulsion liquid chromatography(MELC)[J].J Sep Sci,2005,28(15):2 023.
[7] Simon M Bryant,Kevin D Altira.An initial assessment of the use of gradient elution in microemulsion and micellar liquid chromatography[J].J Sep Sci,2004,27(17-18):1 498.
[8] Eamon M,Sheila D,Joe P,et al.Application of MELC and MEEKC for the analysis of paracetamol and related impurities in suppositories[J].Chromatographia,2008,68(1-2):49.
[9] El-Sherbiny DT,El-Ashry SM,Mustafa MA,et al.Evaluation of the use of microemulsions as eluents in high-performance liquid chromatography[J].J Sep Sci,2003,26(6-7):503.
[10] Malenovic A,Ivanovic D,Medenica M,et al.Retention modelling in liquid chromatographic separation of simvastatin and six impurities using a microemulsion as eluent[J].J Sep Sci,2004,27(13):1 087.
[11] Walash MI,Belal F,EL-Eeany N,et al.Microemulsion liquid chromatographic determination of nicardipine hydrochloride in pharmaceutical preparations and biological fluids application to stability studies[J].J Liq Chromatogr Related Technol,2007,30(8):1 015.
[12] Jancic B,Ivanovic D,Medenica M,et al.Development of liquid chromatographic method for fosinoprilat determination in human plasma using microemulsion as eluent[J].J Chromatogr A,2005,1 088(1-2):187.
