甘草饮片乙酸乙酯提取部位HPLC指纹图谱研究
2011-05-26苏本正孙立立
苏本正, 周 倩, 孙立立*
(1.山东省中医药研究院,山东济南 250014;2.山东中药医药大学,山东济南 250014)
甘草为豆科植物甘草Glycyrrhiza uralensisFischer.、胀果甘草Glycyrrhiza inflateBat.或光果甘草Glycyrrhiza glabraL.的干燥根和根茎[1]。甘草为临床常用中药,是我国2000种草药中用量最大的一味药材[2]。临床上以饮片入药,常见的饮片规格为生品和炙品两种。但是,中药饮片的炮制缺乏标准操作规程的现象目前还相当突出[3-5],需要建立完善的炮制标准来规范它。甘草生品具有补脾益气、清热解毒、祛痰止咳、缓急止痛和调和药性的功效,经蜜炙后增强了其补脾益气的作用,以补脾和胃、益气复脉为主[6-8]。中药炮制后药效改变的根本原因是炮制引起了中药化学成分的变化,前期对甘草及蜜炙甘草的HPLC指纹图谱研究[9]发现,炮制后化学成分的确发生了一定的变化。单独的一两个成分的测定,不能系统、完整地反映甘草的内在质量,目前指纹图谱已成为国际公认的控制中药质量的有效手段[10-12],为进一步追踪变化成分所在部位,本实验提取了甘草乙酸乙酯提取部位,对其进行了指纹图谱研究,并对生、炙品进行了初步比较,从而为进一步明确药效改变的物质基础,解析甘草蜜炙原理提供依据和数据支持。
1 仪器与试药
Waters 2695型高效液相色谱仪,Waters 2996 DAD检测器,1/10万电子天平(Sartorius R200D,德国)。乙腈为色谱纯,水为超纯水,甲醇、乙醇、乙酸乙酯、醋酸均为分析纯。甘草苷对照品(批号11610-200604)购自中国药品生物制品检定所。
甘草饮片:甘草01(产地内蒙古)、甘草02(内蒙古)、甘草03(甘肃)为自制甘草饮片,药材购自山东天一中药饮片有限公司,甘草04(新疆)为自制甘草饮片,药材购自浙江中医药大学中药饮片厂;甘草05购自宁夏银川,产地内蒙古;甘草06购自济南建联大药房,产地内蒙古;甘草07购自南京金陵大药房,产地甘肃;甘草08购自北京圣惠堂中药饮片有限公司,产地内蒙古;甘草09购自济南同仁堂药店,产地内蒙古;甘草10购自山东天一中药饮片有限公司,产地新疆。
炙甘草饮片:炙甘草01、炙甘草02、炙甘草03、炙甘草04分别为甘草01、02、03和04制得蜜炙甘草饮片。炙甘草05购自浙江中医药大学中药饮片厂,产地新疆;炙甘草06购自山东天一中药饮片有限公司,产地新疆;炙甘草07购自济南建联大药房,产地内蒙古;炙甘草08购自北京圣惠堂中药饮片有限公司,产地内蒙古;炙甘草09购自济南同仁堂药店,产地内蒙古;炙甘草10购自南京金陵大药房,产地甘肃。
2 试验方法与结果
2.1 色谱条件 Hyperclone ODS C18柱(4.6 mm×250 mm,5 μm);流动相:乙腈-冰醋酸水溶液(0.2%);柱温:30℃;流速:1 mL/min;检测波长:310 nm。梯度条件见表1。

表1 梯度洗脱程序表Tab.1 Gradient elution
2.2 供试品溶液制备 取(炙)甘草粗粉10 g,加入10倍量水,回流提取3次,每次1 h,滤过,合并滤液,将滤液浓缩至10 mL,加入乙醇使含醇量达到80%,静置24 h,滤过,滤液浓缩至无醇味,继续浓缩至稠膏状,加入适量水使溶解,转移至分液漏斗中,用乙酸乙酯萃取3次,合并萃取液减压回收乙酸乙酯至干,加适量甲醇使溶解,并定容至10 mL,混匀,精密吸取1 mL至25 mL量瓶中,加甲醇定容至刻度,摇匀,经0.45 μm微孔滤膜滤过,即得。
2.3 对照品溶液制备 取甘草苷对照品适量,加甲醇制成每1 mL含20 μg的溶液,即得。
2.4 方法学考察
2.4.1 对照试验 取对照品溶液10 μL,注入高效液相色谱仪,依法测定,记录色谱图。见图1。

图1 甘草苷对照品色谱图Fig.1 HPLC chromatograms of glycyrrhizin
2.4.2 精密度试验 取炙甘草02供试品溶液10 μL,连续进样6次,以甘草苷的保留时间和峰面积为参照,分别对各主要色谱峰相对保留时间和相对峰面积进行统计。结果表明:各色谱峰的相对保留时间和相对峰面积基本一致,RSD均小于3%,符合指纹图谱要求。
2.4.3 稳定性试验 取炙甘草02供试品溶液,在室温下保存,分别于 0,3,6,9,12 h 测定,以甘草苷保留时间和峰面积为参照,分别对各主要色谱峰相对保留时间和相对峰面积进行统计。结果表明,各色谱峰的相对峰面积和相对保留时间基本一致,RSD均小于3%,符合指纹图谱要求。
2.4.4 重复性试验 取炙甘草02饮片粗粉6份,按照供试品溶液的制备方法制成供试品溶液,依次测定,以甘草苷保留时间和峰面积为参照,分别对各主要色谱峰相对保留时间和相对峰面积进行统计。结果表明,各色谱峰的相对保留时间和相对峰面积的RSD均小于3%,符合指纹图谱要求。
2.5 炙甘草饮片乙酸乙酯部位指纹图谱 取各炙甘草样品,照2.2项下方法制备供试品溶液,照上述色谱条件依法进行测定,记录HPLC色谱图。以甘草苷为参照,对各主要色谱峰相对峰面积进行统计,结果见表2。
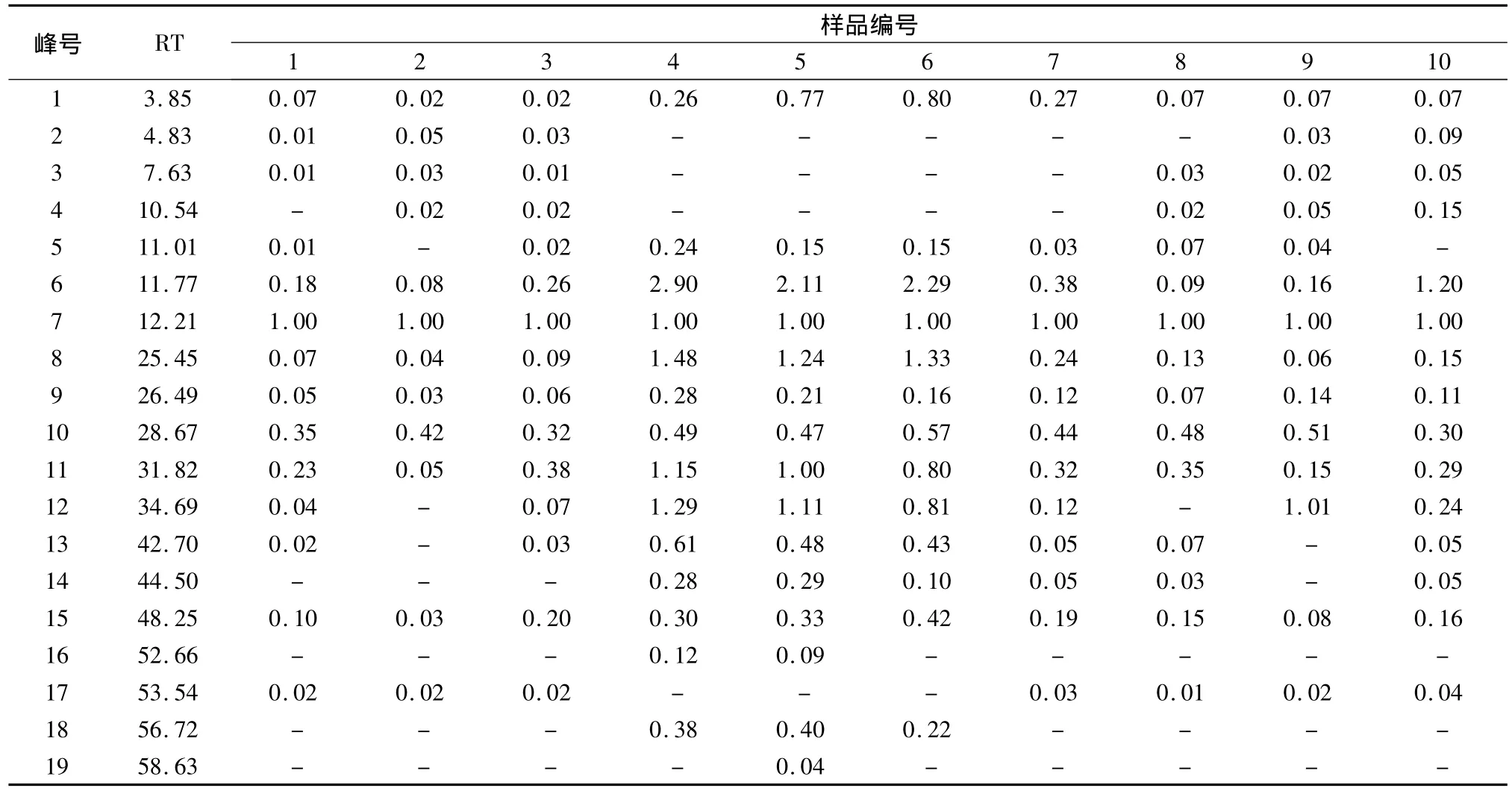
表2 10批炙甘草乙酸乙酯部位指纹图谱(相对峰面积)Tab.2 Relative peak area of HPLC fingerprint common peaks of honey-fried Glycyrrhizae Radix et Rhizoma
炙甘草乙酸乙酯部位共有峰的确定及指纹图谱共有模式的建立 运用“中药色谱指纹图谱相似度评价系统2004A版”对10批炙甘草乙酸乙酯部位指纹图谱进行相似度分析,以S1为参照图谱,经过多点校正、自动匹配,以中位数法,生成对照图谱R,由匹配数据的输出结果得到共有峰共8个,分别为3.85、11.77、12.21、25.45、26.49、28.67、31.82、48.25 min处色谱峰,其中12.21 min峰为甘草苷。除上述色谱峰,在11.01 min左右,除炙甘草02和10外有一共有峰,在34.7和42.70 min左右除炙甘草02和09外有一共有峰,44.496 min除了炙甘草01,02,03和炙甘草09外有一共有峰,56.72 min炙甘草04,05,06有一共有峰,58.63 min炙甘草05较其他甘草多一色谱峰。见图2。
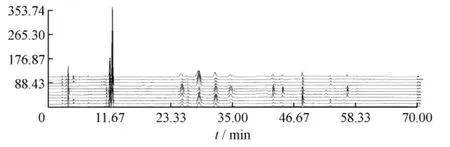
图2 10批炙甘草饮片色谱叠加图(R为对照图谱)Fig.2 The overlapped chart for ten batches of honey-fried Glycyrrhizae Radix et Rhizoma(R is comparison chart)
指纹图谱的相似度评价使用“中药色谱指纹图谱相似度评价系统2004A版”进行相似度分析,结果见表3,经分析,10批次炙甘草饮片与相应对照图谱的相似度均大于0.90,符合要求。

表3 与共有模式比较的蜜炙甘草相似度Tab.3 Results of similarity analysis of honey-fried Glycyrrhizae Radix et Rhizoma
2.6 生甘草饮片乙酸乙酯部位指纹图谱 取各生甘草样品照2.2项下方法制备供试品溶液,照上述色谱条件依法进行测定,记录HPLC色谱图,以甘草苷为参照,对各主要色谱峰相对峰面积进行统计,结果见表4。
生甘草乙酸乙酯部位共有峰的确定及指纹图谱共有模式的建立运用“中药色谱指纹图谱相似度评价系统2004A版”对10批生甘草乙酸乙酯部位指纹图谱进行相似度分析。以S1为参照图谱,经过多点校正、自动匹配,以中位数法,生成对照图谱R,由匹配数据的输出结果得到共有峰共10个,分别为11.01、11.77、12.21、25.47、26.505、28.67、31.81、34.71、42.69、48.24 min。此外,在 17.89min 饮片01、02、06、08、09 有峰,43.97 min 饮片 05 有峰,在44.49 min饮片04、05和10有峰,52.68和56.74 min饮片04和10有峰,58.66 min饮片04有峰。见图3。
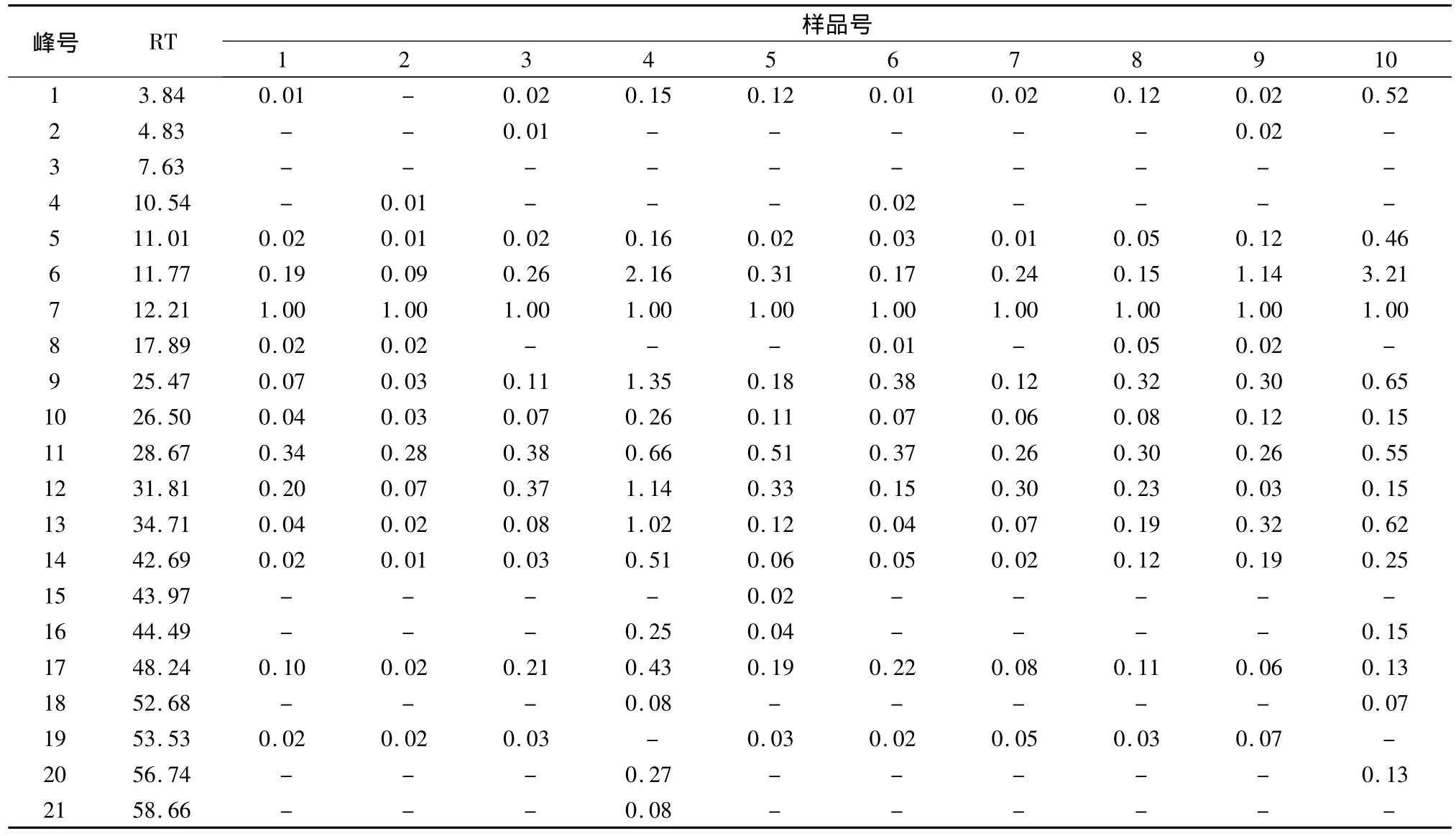
表4 10批甘草乙酸乙酯部位指纹图谱相对峰面积Tab.4 Relative peak area of HPLC fingerprint common peaks of Glycyrrhizae Radix et Rhizoma
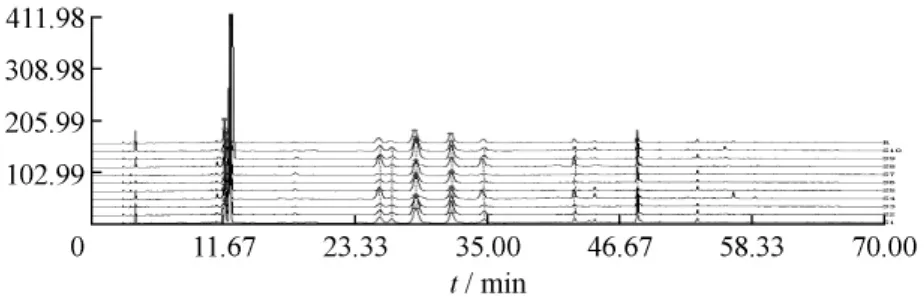
图3 10批生甘草饮片指纹图谱(R为对照图谱)Fig.3 The overlapped chart for ten batches of Glycyrrhizae Radix et Rhizoma(R is comparison chart)
指纹图谱的相似度评价使用“中药色谱指纹图谱相似度评价系统2004A版”进行相似度分析,结果见表5,经分析,10批次生甘草饮片与相应对照图谱的相似度均大于0.90,符合要求。
2.7 生炙饮片乙酸乙酯部位比较 炙甘草01、02、03和04分别由甘草01、02、03和04制得,分别对01,02,03和04甘草炮制前后峰面积进行了统计,比较甘草乙酸乙酯提取部位炮制前后的变化情况,结果见表5。

表5 与共有模式比较的生甘草相似度Tab.5 Results of similarity analysis of Glycyrrhizae Radix et Rhizoma
由表5可见,在四产地甘草共有峰中,10.99、11.74、12.19、25.34、26.42、28.53、34.63、42.67、48.22 min色谱峰经蜜炙后峰面积减小,3.85 min色谱峰经蜜炙后峰面积增加,其余各色谱峰,4.86 min和7.67 min(甘草04无)色谱峰经蜜炙后峰面积增加,10.62 min色谱峰(甘草01和04无),17.89 min(甘草03和04无)、53.53 min(甘草04无)色谱峰经蜜炙后峰面积减小,另甘草04在44.44、52.68、56.74和58.66 min较其他甘草多一色谱峰,峰面积经蜜炙后减小。31.73 min色谱峰炙甘草01和03峰面积略有增加,但与生品相比差异较小,炙甘草02和04峰面积减少,考虑炮制对其影响较小或为积分误差所致。

表5 生炙甘草乙酸乙酯提取部位主要色谱峰峰面积比较Tab.5 Comparison of chemical constituents between Glycyrrhizae Radix et Rhizoma and processed products
3 结论
本实验通过研究建立了甘草饮片乙酸乙酯提取部位的指纹图谱,得到指纹图谱各色谱峰分离度较好,基线平稳,色谱信息较为丰富,而且色谱峰分布均匀,参照峰甘草苷(保留时间12.2 min)与其他色谱峰分离度良好。
通过测定,不同产地甘草饮片与生成的对照图谱比较相似度良好,均大于0.90。各主要色谱峰相对保留时间基本一致,符合指纹图谱的要求。但所含成分有所差异,且峰面积差别较大,说明产地不同其成分之间会有一定的差异。
通过对生炙甘草比较发现,甘草与炙甘草乙酸乙酯部位的化学成分有所不同,各成分的含量比例关系发生了变化,且有新的化学成分产生。11 min之前的色谱峰蜜炙后峰面积普遍增大,11 min之后色谱峰峰面积经蜜炙后降低,说明蜜炙使极性较大成分含量增加,极性较小的成分含量降低。该变化可能为甘草药效作用改变的物质基础。要想明确甘草蜜炙炮制原理,还需进一步对其他提取部位进行分析,同时分离炮制前后变化明显的化学成分,对其进行生理活性筛选,寻找炙甘草药理活性成分,从而为炙甘草炮制工艺和质量标准提供有效的质控指标,全面提高饮片质量,以保证中医临床用药的安全性和有效性提供依据。
[1]中国药典[S].一部.2005:59-60.
[2]刘润堂,马彗英,赵怀生,等.甘草的发展前景及加工技术[J].科研推广,2004,4(4):34-35.
[3]陈吉炎,陈 黎,安志斌,等.中药饮片质控乏力的原因与对策[J].中药材,2003,26(1):43-46.
[4]富同义.中药饮片质量亟待规范[J].中国新医药,2003,2(4):74.
[5]孙 生,齐红梅,李秀霞.中药饮片及其炮制品目前存在的问题[J].天津药学,2001,13(2):30-38.
[6]朱卫星,李爱光,陈 方,等.正交实验优选恒温干燥法蜜炙甘草的工艺研究[J].时珍国医国药,2006,17(8):1407-1408.
[7]王明喜,石志强.生甘草炙甘草临证应用考辨[J].实用中医内科杂志,2005,19(4):383.
[8]崔淑芬,张信青,Lee F S C,等,HPLC指纹图谱应用于炙甘草的炮制研究[J].中成药,2007,29(11):1636-1639.
[9]周 倩,吕 佳,李贵海,等.蜜炙甘草饮片HPLC指纹图谱研究[J].中国中药杂志,2010,35(12):1547-1550.
[10]谢培山.中药色谱指纹图谱鉴别的概念、属性、技术与应用[J].中国中药杂志,2001,26(10):653-655.
[11]韩凤梅,蔡 敏,陈 勇.中药指纹图谱技术研究现状[J].分析科学学报,2004,20(6):647-650.
[12]汪河滨,罗 锋.新疆光果甘草HPLC指纹图谱研究[J].中成药,2008,30(10):1412-1414.
