散发性阿尔茨海默病早老素2基因的新突变
2011-05-25白云峰田菊权文香前田潔
白云峰,田菊,权文香,,前田潔
(1.解放军第302医院中西医结合肝病诊疗与研究中心一科,北京 100039;2.北京大学第六医院生化室,北京 100191; 3.日本神户大学医学系研究科精神神经科,日本 神户 6500017)
阿尔茨海默病(Alzheimer’sdisease,AD)是一种渐进性神经变性疾病,分为家族性AD(FAD)和散发性AD(SAD)两种类型,其中SAD占多数。在欧洲SAD的比例为50%~75%,在日本SAD占80%~90%。目前为止有3种AD的致病基因:β淀粉样蛋白前体(APP)、早老素 1(presenilin-1,PSEN1)和早老素 2(PSEN2)。FAD 大多与 PSEN1、APP 基因突变有关,极少数与PSEN2基因突变有关。迄今为止,世界范围内共发现与FAD相关的PSEN1基因突变有164个,与SAD相关的PSEN1基因突变有3个。与PSEN1基因同源性的PSEN2基因中也有11种基因突变的报道,10种基因突变与FAD相关,1种基因突变与SAD相关。ApoE是SAD和晚发性FAD的易感基因,ApoE等位基因ε4是SAD的主要遗传风险因子[1],然而携带APOEε4的SAD患者并不一定发病,所以可能还存在其他遗传风险因子。我们为了进一步探讨SAD与PSEN2基因突变的相关性,进行了日本人群中PSEN2基因的序列测序。
1 材料与方法
1.1 材料来源
300例患者来自日本神户大学附属医院精神神经科门诊。其中男120例,女180例,年龄56~92岁,平均(72.57±7.56)岁。入组患者均进行常规检查、脑电图、MRI检查,MRI检查显示内颞叶结构萎缩,包括海马结构、杏仁核和内嗅皮质[2,3]。并根据NINCDS/ADRDA统一标准确立为很可能的AD[4],患者或知情者诉有超过6个月的缓慢进行性记忆减退,测试发现有严重的情景记忆损害的客观证据,主要为回忆受损,通过暗示或再认测试不能显著改善或恢复正常,全部患者无阳性家族史。并用简易智能量表(MMSE)进行认知能力评估,评分≤24分者被选入组。对照组300例来自社区随机抽样。其中男120例,女180例,年龄55~94岁,平均(70.97±6.73)岁;进行神经系统检查及MRI检查均正常者被选入组,并且MMSE得分≥28分。此研究经日本神户大学医学伦理委员会批准。
1.2 方法
1.2.1 DNA提取及PCR:提取所有受试者的周围血5 ml,用基因组DNA提取试剂盒从外周血的白细胞中提取基因组DNA。用PCR方法进行PSEN2基因外显子区域的扩增。正向引物及反向引物见表1。用Takara DNA多聚酶进行PCR。反应体系30μL:包括100ng基因组DNA 1μL、30pmol/L的引物各1μL、3μL的 10×PCR 缓冲液、0.2 mmol/L的dNTP 3μL、1U的Takara Taq DNA多聚酶0.15μL。PCR反应条件为:95℃变性9 min后,进入主循环;95℃30 s、各外显子PCR的不同退火温度30 s、72℃1 min共35个循环后72℃延伸10 min。外显子2的PCR退火温度为65℃,外显子4的PCR退火温度为56℃,其他外显子PCR的退火温度为60℃。取PCR产物5μL在2%的琼脂糖凝胶中进行电泳,之后用EB染色并在紫外线下观察结果。
1.2.2 碱基序列测序:采取直接测序PCR产物,首先用PCR纯化试剂盒(Qiagen,USA)纯化25μL的PCR产物,然后进行碱基序列的测序反应(用PCR引物),反应体系为20μL;包括纯化PCR产物1 μL,10 pmol/L的单向引物2μL,BigDye Terminator1.1的4μL,Sequence缓冲液4μL。退火温度为56℃。然后用ABI 3700基因测序仪(ABI,USA)进行测序。核酸序列经数据收集软件整理,用DNAsis2软件进行序列对比分析。
2 结果
采用12对引物(表1),对PSEN2基因的各外显子分别进行PCR及直接测序。测序300例患者组PSEN2基因序列,在3例SAD患者的外显子3上发现了100G>A基因突变,第34号密码子由甘氨酸到丝氨酸(G34S)的变化(图1),而300例对照组中没有发现此基因突变。显然,100G>A基因突变在SAD组中的频率为1%,对照组中的频率为0%。此外,在1例67岁的SAD患者PSEN2基因的外显子4上发现1915C>T基因突变,但此基因突变没有引起氨基酸编码的改变(87H)。
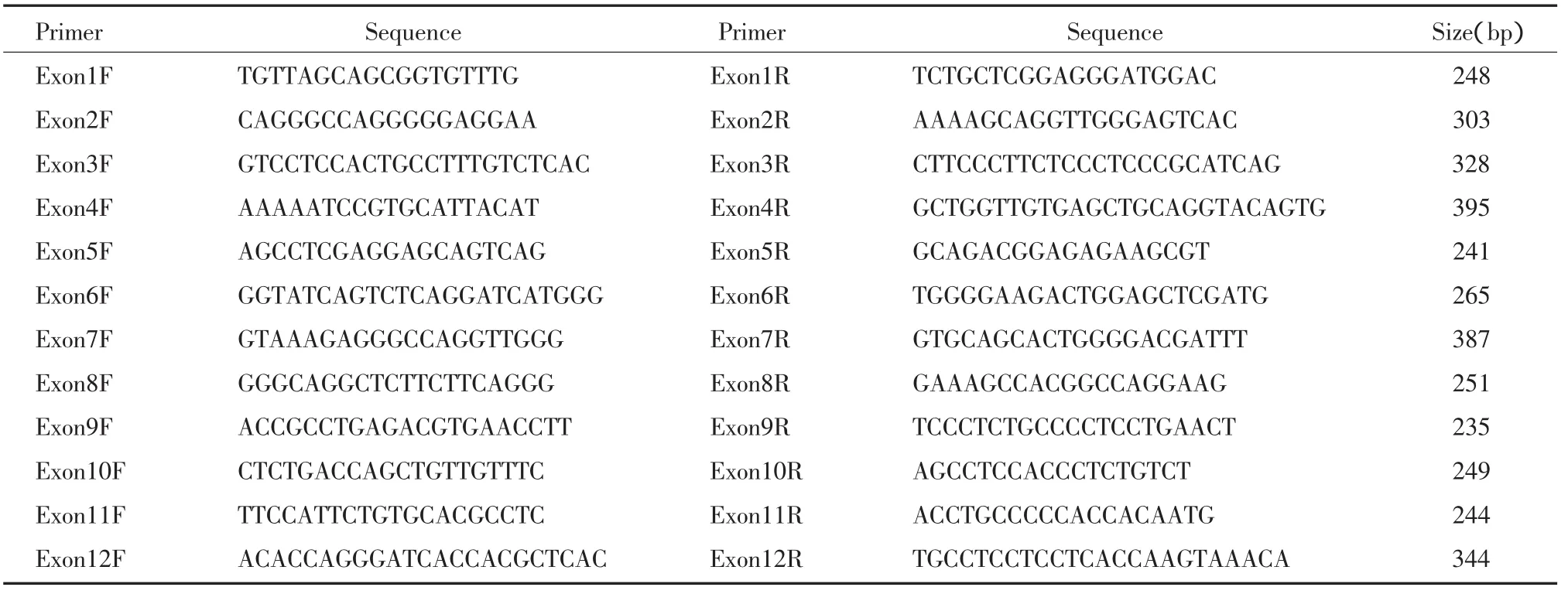
表1 各外显子的引物Tab.1 The primers of exons
3 讨论
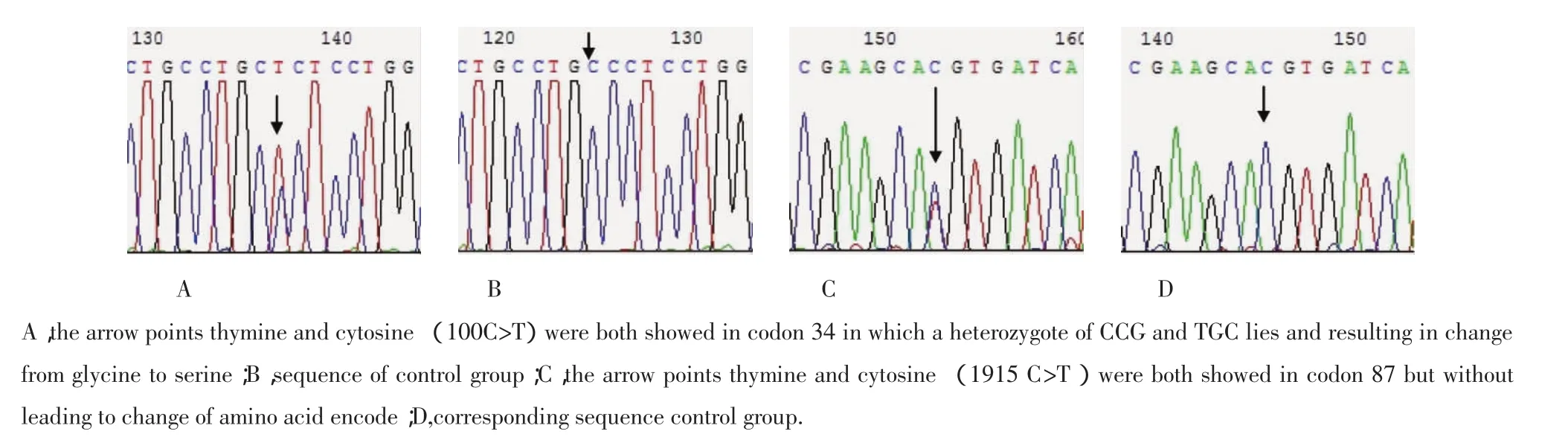
图1 PSEN2基因DNA测序结果Fig.1 The results of DNA sequencing of PSEN2
PSEN2基因全长24 837 bp,位于1号染色体,有12个外显子,从第3个外显子到第12个外显子含有编码区,共2 236 bp。PSEN2蛋白是由PSEN2基因编码的448个氨基酸组成,是8次跨膜的膜蛋白质。PSEN2蛋白与其同源性的PSEN1蛋白是γ分泌酶复合体的组成之一,也是活性中心。PSEN属于天冬酰氨蛋白酶家族(aspartyl protease family),在8个跨膜结构域(transmenbrane domain,TMD)中,2个分别位于TMD6及TMD7中的天冬氨酸残基是催化剪切底物必需的活性位点(图2)。PSEN首先以无活性的全蛋白形式分泌,再经过自身催化的跨膜剪切断裂为功能性片断C端片段(CTF)和N端片段(NTF),才形成有生物功能的PSEN-NTF/CTF二聚体。除此之外,PSEN在C端保守的PAL(prolinealanine-leucine,脯氨酸-丙氨酸-亮氨酸)序列以及在第 4跨膜区中保守的 WNF(tryptophan-asparagine-phenylalanine,色氨酸-天冬酰胺-苯丙氨酸)序列,对于γ分泌酶的活性也很重要[5]。当PSEN蛋白异常时,会改变γ分泌酶的活性,从而导致中枢神经细胞内外产生过量的异常沉积的Aβ42,或导致 Aβ42/Aβ40 的比例失调[6]。目前PSEN2基因测序的研究中,已发现11个PSEN2基因突变,所有基因突变的表现型均为AD。
为了探索AD的致病基因,以微阵列或SNP(singlenucleotidepolymorphism)为标记检测碱基全序列的研究是目前国内外的热门。根据国际人类基因组计划已经清楚人类的全部基因,并进行了大量的基因分析,尽管这样还有很多碱基替换的报道接连不断。1995年,Sherrington等[7]发现 M146L、H163R、A246E、L286V和C410Y 5个位于PSEN1基因的突变。相继100多个PSEN1基因突变在家族性AD中陆续被报道,其中3个PSEN1基因突变是在SAD中被报道。其一是Dumanchin等[8]在法国的37岁SAD患者中发现的M139K基因突变。其二是Kamimura等[9]在日本的散发人群中发现的1例具有H163R基因突变的AD患者。这两个基因突变分别位于PSEN1蛋白CTF第5外显子的TM-II和第6外显子的HL-II,认为均具有致病性[10]。其三是Scacchi等[11]在85岁SAD患者的外显子4上发现的N32(AAT>AAC)的基因突变。该基因突变位于PSEN1蛋白的非功能区NTF,认为不具有致病性。另外,Rogaeva等[12]在家族性AD中发现了R35Q、A79V基因突变。这两种基因突变也位于PSEN1蛋白的非功能区NTF,认为与AD发病无关。
PSEN2基因分析中,Bird首次提出PSEN2基因突变会引起家族性AD。Bird报道的Volga German家系的发病原因为N141I的基因突变,其AD的平均发病年龄为58.7岁[13]。同期,Rogaev等在意大利FLO10家系中发现了具有PSEN2基因M239V基因突变的9例患者,发病年龄为45~88岁,平均50岁。可见PSEN2基因点突变有早发性AD也有晚发性AD,且外显率不是100%,同一家族里具有PSEN2基因点突变的成员不一定发病。PSEN2基因突变在SAD人群中也有报道,Cruts等在荷兰的SAD人群中发现了R62H基因突变,位于PSEN2蛋白的非功能区NTF,患者的发病年龄为62岁,在SAD中此基因突变的频率为1%,正常对照组中的频率为0%。Walker等[14]将PSEN2基因突变载体转染到神经细胞内,观察Aβ的含量。发现N141I、M239V、M239I和T122P等基因突变体的Aβ42或者Aβ42/Aβ40的比例比对照组有明显增多,但R62H、S130L、V148I、D439A 等基因突变体没有相应的变化。可见部分PSEN2基因突变可能是与AD发病无关的点突变。本文的G34S基因突变与R62H同样在SAD中被发现,同样位于PSEN2蛋白的非功能区NTF,G34S基因突变在SAD中的频率为1%,在对照组中频率为0%,曾被认为有可能与AD的发病有关[15]。G34S基因突变可能与SAD发病无关的正常多态性,但其生物学功能有待于进一步探讨。对于1915 C>T(87H)的基因突变没有引起氨基酸编码的改变,且同样位于PSEN2蛋白非功能区NTF,所以不会影响PSEN2蛋白的功能。
今后为了更多的发掘与AD相关的新的基因或者新的基因突变,有必要继续进行基因分析。在AD发病中除了显性遗传之外,还有多个基因的多重作用,并且在明确的家系中也存在非显性遗传方式遗传、或者母性遗传方式遗传的报道。更不应忽视人种差异、区域、环境、放射线等基因以外的影响。AD遗传学不像单一的显性遗传那么简单,应该是复合因子相互影响的一个复杂的问题。
[1]Saunders AM,Strittmatter WJ,Schmechel D,et al.Association of apolipoprotein Ealleleepsilon 4 with late-onset familial and sporadic Alzheimer’sdisease[J].Neurology,1993,43(8):1467-1472.
[2]Jack CR Jr,Petersen RC,Xu YC,et al.Medial temporal atrophy on MRIin normal aging and very mild Alzheimer’sdisease[J].Neurology,1997,49(3):786-794.
[3]母其文,谢敬霞,翁雅琴,等.早期阶段Alzheimer病内颞叶记忆系统多相关结构多变量的定量MRI研究[J].中华放射学杂志,1998,32(12):807-811.
[4]McKhann G,Drachman D,Folstein M,et al.Clinical diagnosis of Alzheimer's disease:report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease[J].Neurology,1984,34 (7):939-944.
[5]Tomita T,Watabiki T,Takikawa R,et al.The first proline of PALP motif at the Cterminus of presenilins is obligatory for stabilization,complex formation,and gamma-secretase activities of presenilins[J].JBiolo Che,2001,276(35):33273-33281.
[6]Joanna LJ,Daniel JF,Jeffrey A,et al.Mutant presenilins specifically elevate thelevelsof the Aβ42 residue β-amyloid peptide in viva evidencefor augmentation of Aβ42-specific γsecretase[J].Human Mol Genet,2004,13(2):159-170.
[7]Sherrington R,Rogaev EI,Liang Y,et al.Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease[J].Nature,1995,375(6534):754-760.
[8]Dumanchin C,Brice A,Campion D,et al.Denovopresenilin 1 mutations are rare in clinically sporadic,early onset Alzheimer's disease cases.French Alzheimer’s Disease Study Group [J].J Med Gen,1998,35(8):672-673.
[9]Kamimura K,Tanahashi H,Yamanaka H,et al.Familial Alzheimer’s diseasegenesin Japanese[J].JNeuro Sci,1998,160(1):76-81.
[10]Cruts M,Van BC.Presenilin mutations in Alzheimer’s disease[J].Hum Mut,1998,11(3):183-190.
[11]Scacchi R,Gambina G,Moretto G,et al.A mutation screening by DHPLC of PSEN1 and APP genes reveals no significant variation associated with the sporadic late-onset form of Alzheimer’s disease[J].Neuro Lett,2007,418(3):282-285.
[12]Rogaeva EA,Fafel KC,Song YQ,et al.Screening for PS1 mutations in a referral-based series of AD cases:21 novel mutations[J].Neurology,2001,57(4):621-625.
[13]Bird TD,Lampe TH,Nemens EJ,et al.Familial Alzheimer’s disease in American descendants of the Volga Germans:probable genetic founder effect[J].Ann Neur,1988,23(1):25-31.
[14]Walker ES,Martinez M,Brunkan AL,et al.Presenilin 2 familial Alzheimer’sdisease mutations result in partial loss of function and dramatic changes in Abeta 42/40 ratios[J].JNeuroche,2005,92(2):294-301.
[15]Yasuda M,Quan W,Maeda K,et al.Genetic Analysis of Japanese Cases with Alzheimer Disease[J].Ann Re Pharma Res Fou,2004,36:242-247.
