微波辅助合成咪唑类化合物的研究
2011-05-18孟江平徐强宋仲容陈凤鸣何家洪
孟江平,徐强,宋仲容,陈凤鸣,何家洪
(1.重庆文理学院化学与环境工程学院,重庆 永川 402168;2.重庆市高校微纳米材料工程与技术重点实验室,重庆 永川 402160)
微波在化学中的应用开辟了微波化学这个化学新领域[1].微波可以与化学体系发生作用从而促进各类化学反应的进行.它对化学反应作用是非常复杂的,一方面是反应物分子吸收了微波能量,提高了分子运动速度,致使分子运动杂乱无章,导致熵的增加;另一方面,微波对极性分子的作用,迫使其按照电磁场作用方式运动,每秒变化2.45×109次,导致了熵的减小.此外,它还存在一种不是由温度引起的非热效应.微波作用下的有机反应,改变了反应动力学,降低了反应活化能.近年来,大量的实验已经证实,微波可以极大地提高一些化学反应的反应速率,使一些在通常条件下不易进行的反应迅速形成.传统上,有机合成或药物合成采用外部热源进行传导加热,如油浴或加热套等.但是传统的加热方法多是间接加热,并受制于对流和必须透过的各种材料的导热性,相对而言它在向体系传输能量时速度缓慢,效率不高,而且导致反应容器的温度高于反应混合物的温度.此外,被加热底物内部会产生温度梯度,而且反应物局部过热也可导致产物、底物或者试剂的分解.相反,通过微波能量和反应混合物中的分子如溶剂、试剂或催化剂的直接耦合,可以直接进行充分的内部加热[2].
咪唑环是生物体内组胺、组氨酸产生生物活性和发挥生理作用的重要基团[3-5].咪唑环是结构中含有2个氮原子的五元芳氮杂环,易产生多种非共价键相互作用,如氢键、与金属离子配位和π-π作用等[6-8].以这种特殊结构的咪唑环构筑的咪唑类衍生物具有较大的发展潜力,如作为人工受体用于分子识别[9],作为人工酶用于仿生催化[10],作为药物具有广泛的生物活性,如组胺受体拮抗剂[11]、酶抑制剂[12]、质子泵抑制剂[13]、抗微生物[14]、抗癌[15]等等.因此,近年来咪唑类化合物的合成与开发越来越受到人们的重视.本文结合自己的研究工作,就近年来有关微波辐射合成咪唑类化合物的国内外相关情况进行综述.
1 微波辅助1,2-二酮和醛合成咪唑类化合物
通过1,2-二酮与醛及其衍生物的反应合成咪唑及其类似物是一个相当传统的合成路径.传统的方法往往需要较为苛刻的条件——强酸性条件,较高的温度,很长的时间等.近年来,随着微波技术在有机合成中的广泛应用,极大地改善了用1,2-二酮与醛及其衍生物合成咪唑的条件.微波辐射1,2-二酮与醛合成咪唑具有速度快、产率高、污染少、安全性高等优点.
Wolkenbergd等[16]在乙酸铝的存在下,由 1,2-二酮和醛简单而高产率的合成了2,4,5-三取代咪唑3.微波照射,温度为180℃,在乙酸铝的存在下,5min之内,1,2-二酮和醛缩合合成烷基、芳基和杂芳基取代的咪唑衍生物,产率可达76%~99%.在三乙胺碱性条件下,2,4,5-三甲基咪唑与苄氯进一步进行微波辅助烷基化反应,以43%的总产率得到了生物碱勒皮啶(lepidine)4.带有不对称咪唑鎓结构的勒皮啶4在微摩尔级显示了抗多种人肿瘤细胞株的细胞毒性,该化合物作为临床抗肿瘤药物具有进一步开发价值.值得注意的是,中间体2,4,5-三甲基咪唑的制备在技术上简便、快速,与以前的路径相比产率更高.化合物3和4的合成路线如图1所示.

图1 化合物3和4的合成路线
2 微波辅助酮肟和醛合成咪唑类化合物
Sparks和Combs等[17]研究了以非对称酮肟与不同的醛为原料,利用微波辅助合成咪唑类化合物的方法,按照这一方式高产率地合成了N-羟基咪唑7.随后以三氯化钛为催化剂,将其定量还原为咪唑化合物8,反应时间仅需5min.研究发现,在高反应温度如200℃下处理酮肟与醛及乙酸铵时,经原位切断热易变的N—O键直接得到了目标化合物8.利用这种优化的条件可制备多样化、高效率的2,4,5-三(杂)芳基咪唑,给咪唑环的合成带来了新的方法.化合物7和8的合成路线如图2所示.

图2 化合物7和8的合成路线
3 微波辅助多组分反应(MCRs)合成咪唑类化合物
多组分反应(Multicomponent Reactions,MCRs)是指3个或3个以上的起始原料进入反应,用“一锅煮”的方法最终生成一个终产物,在终产物结构中含有所有原料片段的合成方法[18].多组分反应由于其高度的原子经济性、收敛性、出色的产率等特性以及在药物研究中先导物的发现和结构优化等方面的重要应用,在有机和药物化学领域发挥着越来越重要的作用.应用多组分反应构筑新的、具有各种生理活性的杂环化合物已成为杂环合成的重要手段之一[19-20].
Ugi四组分缩合反应,即胺、醛和酮、羧酸以及异腈结合产生α-酰基氨的反应是目前有机合成中的常用方法,因其通过不同的起始原料可以获得范围很宽的产物而具有特别意义.在以三氟甲磺酸钪为催化剂进行的Ugi型三组分缩合反应中,杂环脒与醛和异腈在室温下反应通常需要长达72 h的反应时间才能生成期望的稠合3-氨基咪唑.Ireland等[21]在微波条件下合成了咪唑类化合物12,合成路线如图 3所示.在160℃下以甲醇作为溶剂,在有些例子中采用乙醇为溶剂,10min就可以产生与室温相同过程类似的产率,而时间只是室温反应的一小部分,极大地提高了合成效率,缩短了反应时间.

图3 化合物12的合成路线
在乙酸铵存在下,醛、2-氧代硫代乙酰胺以及溴代烷之间的具有多种包容性三组分缩合合成咪唑类化合物[22].利用微波辅助多组分反应原理,以结构不同的醛,溴代烷和2-氧代硫代乙酰胺为原料,合成了咪唑类化合物16,合成路线如图4所示.生物活性研究表明,16是一类潜在的酰基辅酶A/胆固醇酰基转移酶抑制剂、止痛剂以及血管紧张素受体的拮抗剂.
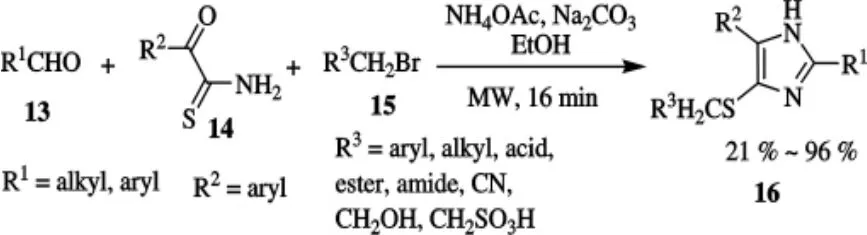
图4 化合物16的合成路线
4 微波辅助合成咪唑啉及其衍生物
咪唑啉是咪唑的重要衍生物之一,在医药、催化剂、洗涤品及缓蚀剂等领域具有广泛用途.传统的合成方法是采用不同种类的羧酸、酯、腈或酰氯等与乙二胺或多胺类化合物反应制得.这一传统的方法具有耗时长、产率低、分离困难及产物成分复杂等缺点.微波辅助技术极大地改善了这些缺点,为咪唑啉类化合物的高通量合成带来了光明的前景.
刘金艳等[23]以吡啶2,6-二甲酸与乙二胺为原料,合成了吡啶咪唑啉类化合物20,合成路线如图5所示.这一合成方法显著缩短了反应时间,提高了产率,减少了环境污染.反应过程中,微波功率为450 W,辐射时间为65min时产率最好.
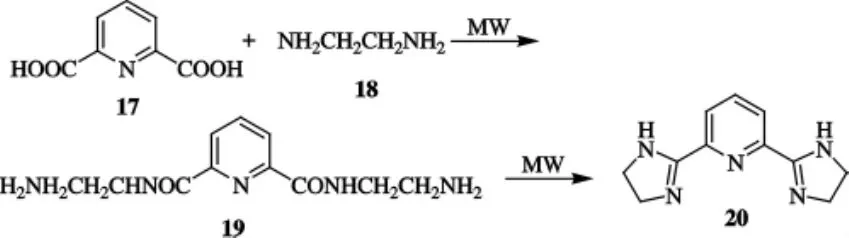
图5 化合物20的合成路线
Merriman等[24]报道了由固相方法得到N-酰基-1,2-二芳基-1,2-乙烷二胺的环化,通过在二氯甲烷溶液中以三甲硅基多聚磷酸盐(TMS-PP)处理后,生成了4,5-二芳基咪唑啉22,合成路线如图6所示.由微波在140℃下加热8 min获得了最佳结果.以这种方法制备了含有38个化合物的库,产生了一个新且有效的P2X7受体拮抗剂家族.

图6 化合物22的合成路线
在氧化锌的存在下,以DMF为溶剂,在微波照射120℃下亚乙基二胺和脲进行缩合,以95%的分离产率得到了咪唑啉-2-酮25,产率高达95%,合成路线如图7所示.这一方法成功的关键之处在于,反应需要在负压下完成,以便从反应混合物中除去形成的氨.这一方法也适用于不同的二胺与氨基醇的反应.

图7 化合物25的合成路线
5 微波辅助合成苯并咪唑及其衍生物
通过邻苯二胺与羧酸及其衍生物的反应合成苯并咪唑及其衍生物是一个相当传统的合成路径,传统的方法往往需要较为苛刻的条件——强酸性条件、较高的温度、很长的时间等.近年来,随着微波技术在有机合成中的广泛应用,极大的改善了用邻苯二胺与羧酸合成苯并咪唑的条件.微波辐射邻苯二胺与羧酸合成苯并咪唑具有速度快、产率高、污染少、安全性高等优点.
Vanelle等[25]研究发现,微波辐射可以有效地促进α-羟基羧酸与邻苯二胺的反应合成2-羟甲基苯并咪唑,其反应效率较传统加热方法有明显提高.传统加热合成时,需要120 h,而采用微波辐射时,反应仅仅需要1.5 h就能够高收率合成2-羟甲基苯并咪唑26,合成路线如图8所示.

图8 化合物28的合成路线
以邻苯二胺和醛为原料合成苯并咪唑也是一个较为传统的方法,这一传统方法存在反应时间较长、副反应复杂、分离提纯困难等缺点.将传统的加热方式改为微波辐射,将会更好地改善合成步骤、缩短反应时间、降低生产成本等.Jacob等[26]在微波辐射下,用 SiO2/ZnCl2作催化剂,合成了1,2-双取代苯并咪唑31,合成路线如图9所示.反应时间为1.5min,收率较高.其中,R为苯环时,用传统的加热方式,温度为75℃,收率为65%,而利用微波辐射反应,温度降低为65℃,收率可达92%.
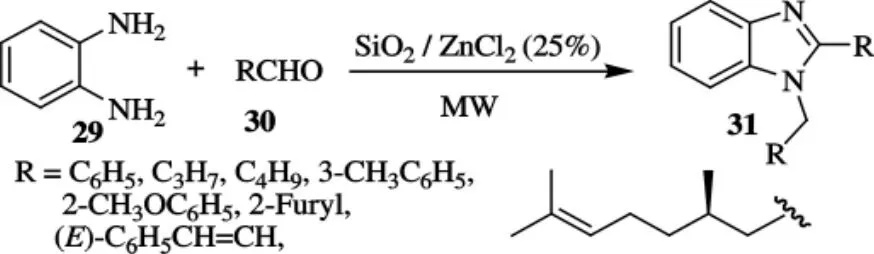
图9 化合物31的合成路线
6 结语
综上所述,微波辐射合成咪唑类化合物较传统的合成方法有以下优点:一是使咪唑的合成从传统的污染性、有毒性逐渐走向“绿色合成”;二是极大地缩短了反应的时间,产率更高,反应外观更加干净;三是对于某个给定的咪唑合成反应,溶剂的选择不再取决于沸点高低,而更倾向于反应介质的介电能力;四是由于微波辐射合成周期短,为未来咪唑药物分子的筛选提供了广阔的分子库.因此,采用微波辐射技术,给咪唑的合成带来了简洁、方便的途径,也为未来咪唑合成方法提供了光明的前景.我们相信,随着微波技术的不断发展,咪唑的微波合成将会得到快速发展,这将为寻找结构新颖、生物活性良好的咪唑类药物中间体提供更加广阔的选择空间.
[1]C O卡帕,A斯塔德勒.微波在有机和医药化学中的应用[M].麻远,译.北京:化学工业出版社,2007:206-209.
[2]Yoon J,Kim S K,Singh N J,et al.Imidazolium receptors for the recognition of anions[J].Chem Soc Rev,2006,35(4):355-360.
[3]周成合,张飞飞,甘淋玲,等.超分子化学药物研究[J].中国科学B辑:化学,2009,36(3):208-252.
[4]Zhou C H,Gan L L,Zhang Y Y,et al.Review on supermolecules as chemical drugs[J].Sci China Ser B:Chem,2009,52(4):415-458.
[5]Zhang F F,Gan L L,Zhou C H.Synthesis,antibacterial and antifungal activities of some carbazole derivatives[J].Bioorg Med Chem Lett,2010,20(6):1881-1884.
[6]Wang X L,Wan K,Zhou C H.Synthesis of novel sulfanilamide-derived 1,2,3-triazoles and their evaluation for antibacterial and antifungal activities[J].Eur J Med Chem,2010,45(10):4631-4639.
[7]孟江平,耿荣霞,周成合,等.苯并咪唑类药物研究进展[J].中国新药杂志,2009,18(16):1505-1514.
[8]袁勇,周成合,刘嫱,等.新型合成抗菌药物研究新进展[J].中国新药杂志,2007,16(5):343-349.
[9]Jiang H Y,Zhou C H,Luo K,et al.Chiral imidazole metalloenzyme models:Synthesis and enantioselective hydrolysis for α -amino acid esters[J].J Mo Cat A:Chem,2006,260(1-2):288-294.
[10]吴俊,米佳丽,周成合.组胺H3受体配体研究进展[J].中国药学杂志,2007,42(6):404-409.
[11]蔡佳利,李硕,周成合,等.咪唑类抗癌药物研究进展[J].中国新药杂志,2009,18(7):21-31.
[12]孟江平,卢一卉,海力且木·依不那音,等.苯并咪唑类酶抑制剂研究进展[J].中国生化药物杂志,2008,29(6),422-425.
[13]孟江平,于克贵,甘淋玲,等.大环类超分子药物研究[J].西北师范大学学报:自然科学版,2008,44(suppl):242-245.
[14]孟江平,于克贵,周成合.2,4-二氯苄类苯并咪唑环番的合成[J].有机化学,2007,27(suppl):557.
[15]Zhou C H,Zhang Y Y,Yan C Y,et al.Recent researches in metal supramolecular complexes as anticancer agents[J].Anti-Cancer Agents in Med Chem,2010,10(5):371-359.
[16]Wolkenberg S E,Wisnoski D D,Lindsler W H,et al.Efficient synthesis of imidazoles from aldehydes and 1,2-diketones using Microwave irradiation [J].Org Lett,2004,6(9):1453-1456.
[17]Sparks R B,Combs A P.Microwave-assisted synthesis of 2,4,5-triaryl-imidazole;A Novel Thermally Induced N-Hydroxyimidazole N—O bond cleavage[J].Org.Lett,2004,6(14):2473-2475.
[18]朱映光,瞿昌伟,胡文浩.不对称多组分反应[J].有机化学,2010,22(7):1380-1396.
[19]Sha F,Huang X.A multicomponent reaction of arynes,isocyanides,and terminal alkynes:Highly chemo-and regioselective synthesis of polysubstituted pyridines and isoquinolines[J].Angew Chem Int Ed,2009,48(19):3458-3461.
[20]陈育兰,严胜骄,林军.多组分反应在杂环合成中的应用[J].广东化工,2010,37(3):4-6.
[21]Ireland S M,Tye H,Whittaker M.Microwave-assisted multi-component synthesis of fused 3-aminoimidazoles[J].Tetrahedron Lett,2003,44(23):4369-4371.
[22]Coleman C M,Mac Elroy J M D,Gallagher J F,et al.Microwave parallel library generation:Comparison of a conventional-and microwave-generated substituted 4(5)-sulfanyl-1H-imidazole library[J].J Comb Chem,2002,4(1):87-93.
[23]刘金艳,肖小明,谭年元,等.微波辐照2,6-二(2'-咪唑啉-2'-基)吡啶的合成及其与DNA的相互作用[J].应用化学,2010,27(6):658-663.
[24]Merriman G H,Ma L,Shum P,et al.Synthesis and SAR of novel 4,5 diarylimidazolines as potent P2X7receptor antagonists [J].Bioorg Med Chem Lett,2005,15(4):435-438.
[25]Boufatah N,Gellis A,Maldonado J,et al.Efficient microwave-assisted synthesis of new sulfonylbenzimidazole-4,7-diones:heterocyclic quinones with potential antitumor activity[J].Tetrahedron,2004,60(41):9131-9137.
[26]Jacob R G,Dutra L G,Radatz C S,et al.Synthesis of 1,2-disubstitued benzimidazoles using SiO2/ZnCl2[J].Tetrahedron Lett,2009,50(13):1495-1497.
