酒精性肝病的发病机制:实质细胞与非实质细胞间的相互作用
2011-05-15JessicaCOHENLauraNAGY
Jessica I COHEN* Laura E NAGY*†
Departments of*Pathobiology,†Gastroenterology and Hepatology,Cleveland Clinic,and‡Department of Nutrition,Case Western Reserve University,Cleveland,Ohio,USA
ALD
长期摄入酒精将导致肝损伤。肝损伤逐级进展,从酒精性脂肪肝、酒精性脂肪性肝炎直至肝纤维化和肝硬化,每一阶段均有其独特的病理学特征。脂肪肝或称脂肪变性,是肝损伤最初期的表现,存在于约90%的饮酒者中,其标志为肝细胞内脂质积聚和肝肿大。约20%的酗酒者将进展至肝损伤的
酒精与公共卫生
酗酒与包括肝炎、肝硬化和胰岛素抵抗在内的60余种疾病有关,是全球性健康问题的主要成因之一。据估计,美国约有1800万酗酒者和1000万以上的酒精性肝病(ALD)患者,年酗酒相关医疗费用高达1660亿美元。根据美国国立酒精滥用与酒精中毒研究所的资料,2004年约2.8万美国居民死于酒精性肝硬化,约40%的致死性车祸与饮酒有下一阶段——脂肪性肝炎。该阶段以炎症反应、肝细胞坏死和凋亡、ALT、AST等血清肝酶升高为标志。酒精性肝炎的临床表现不一,轻者以实验室检查见转氨酶轻度升高为惟一征象,重者肝功能严重异常,出现黄疸、肝性脑病、腹水、食管静脉曲张、凝血障碍以至昏迷等并发症。如停止摄入酒精,脂肪肝和轻度脂肪性肝炎是可逆的,重度酒精性脂肪性肝炎的死亡率则高达30%~60%。
随着肝损伤的进展,约半数酒精性肝炎患者肝星状细胞(HSC)活化并产生过量胶原,发生肝纤维化和肝硬化。在此阶段,细胞外基质(ECM)过度沉积,导致肝窦丢失、肝硬化,部分患者可致死。ALD患者进展至肝硬化的比例不一,不同研究报道为10%~30%。酒精性肝硬化仍为全球肝移植最常见的指征之一。
ALD的起始和进展机制
“二次打击”假说是酒精性肝损伤进展的可能机制之一,第一次打击为乙醇暴露导致肝脂肪变性。长期乙醇暴露对肝脏的脂质内环境稳态有重大影响,引起脂肪酸氧化减少、合成增加,三酰甘油从肝脏输出受抑,这些脂质代谢的变化最终导致脂肪变性。第二次打击是继续暴露于乙醇的结果,如乙醇代谢使氧化应激增加。此外,与肥胖症或病毒性肝炎等共病亦可作为第二次打击加剧酒精性肝损伤。
与“二次打击”假说相似,“致敏和初始”学说认为摄入酒精可致敏肝脏,使之对次级危险因素引起的损伤更为易感。乙醇代谢对肝细胞功能的影响可能是肝脏致敏的成因。“初始”指诱导或激活肝损伤机制的早期事件,如长期乙醇暴露后,肝内驻留型巨噬细胞Kupffer细胞对内毒素变得敏感,诱导产生促炎细胞因子和活性氧簇(ROS),导致肝损伤。
乙醇的代谢机制
乙醇主要在肝脏内由乙醇脱氢酶(ADH)代谢生成乙醛,同时辅酶Ⅰ(NAD)经还原反应转变为还原型辅酶Ⅰ(NADH)(见图1)。绝大多数乙醇经由此途径代谢。血液中ADH含量很低,约为0.2~2 mmol/L,乙醇浓度亦很低。毒性代谢产物乙醛能诱导肝脏氧化应激,损伤线粒体功能。NADH产生增加可打破肝脏的氧化-还原平衡,干扰糖异生。乙醛由乙醛脱氢酶代谢生成乙酸,乙酸作为三羧酸循环的底物可产生能量。
乙醇代谢的第二条主要途径为微粒体乙醇氧化系统,该途径由细胞色素P4502E1(CYP2E1)催化(见图1)。与ADH相似,CYP2E1亦主要存在于肝细胞中,然而个别研究报道长期乙醇暴露后Kupffer细胞中CYP2E1表达增加。由于CYP2E1的含量高达10~15 mmol/L,因此该途径的作用为代谢过量的乙醇。长期乙醇暴露后,人类和啮齿类动物肝内的CYP2E1含量增高4~10倍。在动物和细胞模型中,乙醇暴露在体内和体外均可增加CYP2E1的合成并减少其降解。除乙醇外,CYP2E1还能代谢对乙酰氨基酚等药物、麻醉剂和工业溶剂。乙醇经CYP2E1代谢产生ROS,ROS可与蛋白质和脂类作用,导致氧化损伤。
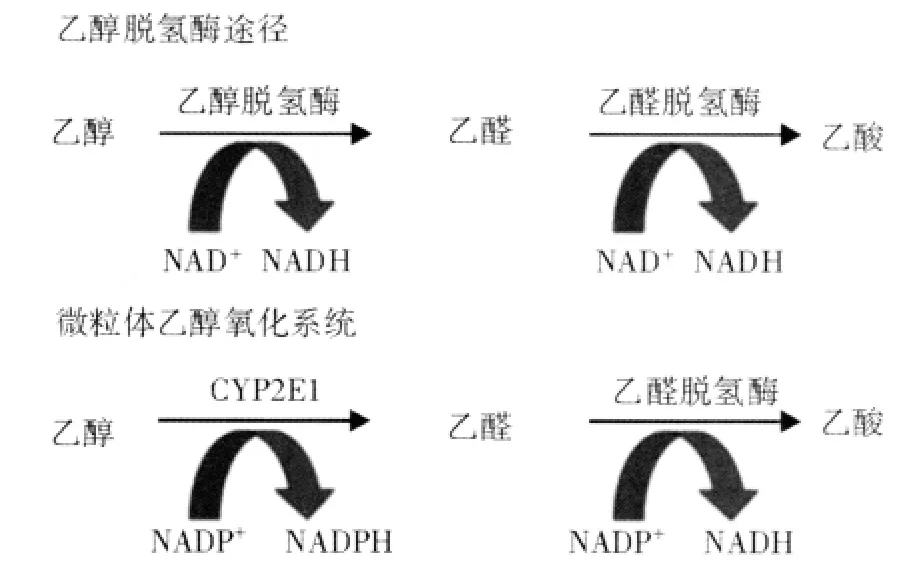
图1 肝脏乙醇代谢的主要途径
乙醇代谢的第三条途径为非氧化途径,该途径系由脂肪酸乙酯(FAEE)合酶介导产生FAEEs。FAEEs是乙醇和脂肪酸的酯化产物,具有细胞毒性。滥用酒精最常损伤肝脏和胰腺,这两个器官中FAEE和FAEE合酶浓度最高。由于停止饮酒后循环血液中的FAEE可以被清除,血清FAEE可作为急性和慢性酒精摄入的临床标记物。
乙醇对肝内细胞的作用
肝细胞细胞器应激:肝细胞是肝内乙醇毒性作用的主要靶点。长期乙醇暴露可损伤肝内多种代谢和解毒路径的功能,对许多细胞器的活性亦有重大影响(见图2)。肝内乙醇毒性作用的关键靶点之一为线粒体,长期乙醇暴露能在多重水平损伤线粒体功能,导致生物能量减少、ROS产生增加、线粒体DNA损伤和蛋白质合成抑制、谷胱甘肽运输异常以及对线粒体通透性改变的敏感性增加。长期乙醇暴露还可致内质网应激和蛋白酶体活性降低。肝细胞内的囊泡转运被破坏,伴高尔基体运输和受体介导的胞吞作用受损,这些改变可能是由微管功能破坏所致。近期研究显示,长期乙醇暴露还可使组蛋白乙酰化的精细调控发生紊乱。上述由乙醇引起的肝细胞细胞器应激综合作用,使肝细胞的代谢和解毒活性受损,并通过坏死和凋亡通路使之对细胞死亡更为敏感。
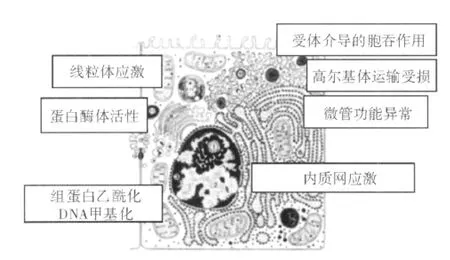
图2 肝细胞内乙醇作用的靶细胞器
肝窦内皮细胞结构改变:肝窦内皮细胞是一组特殊的内皮细胞,其特征性的窗孔结构使大分子物质和红细胞易于流入Disse间隙。除已明确乙醇暴露可致窗孔结构丢失外,目前关于长期乙醇暴露对肝窦内皮细胞的直接和(或)间接作用所知甚少。
免疫反应在ALD起始和进展中的作用:免疫反应由先天性和获得性免疫系统两部分组成,两者在机体对损伤或应激产生应答的多个阶段中均有密切联系。先天性免疫反应的组分,包括自然杀伤(NK)细胞、NK-T 细胞、Kupffer细胞和补体系统,以及T细胞和抗体依赖性获得性免疫反应,参与了肝脏对不同类型损伤,如细菌和病毒感染、毒素(如乙醇)暴露、肝部分切除术和缺血-再灌注等的应答。目前对Kupffer细胞在损伤应答中的作用了解最为深入。长期乙醇暴露可降低NK细胞活性。NK细胞能于早期活化阶段杀伤HSC,并通过释放含粒酶、穿孔蛋白、死亡配体如肿瘤坏死因子相关凋亡诱导配体(TRAIL)和多种促炎细胞因子的颗粒,充当杀伤病毒感染细胞时的重要效应细胞。而NK-T细胞则于对乙醇产生应答的早期被激活,在肝脏先天性免疫反应的防御机制中起关键作用。
肝脏驻留型巨噬细胞Kupffer细胞活化和敏感性增强:长期暴露于乙醇可使空肠共生细菌过度生长。微生物群的增加以及伴随发生的乙醇所致的小肠屏障功能破坏,促使革兰阴性细菌易位进入门静脉循环。予大鼠和小鼠酒精灌胃或人类摄入酒精后,血中内毒素浓度均增高。予大鼠抗生素治疗以清除革兰阴性细菌,酒精的肝损伤作用减弱。
门静脉血流通过第一级毛细血管床将内毒素或脂多糖(LPS)带入肝脏内。而Kupffer细胞则发挥解毒作用,将LPS清除出血液循环。LPS激活Kupffer细胞,产生细胞因子如肿瘤坏死因子-α(TNF-α)等炎症介质和ROS。Kupffer细胞对ALD的进展具有关键意义。予大鼠巨噬细胞毒素氯化钆治疗可消除Kupffer细胞,预防乙醇诱导的肝损伤。LPS介导的信号亦为乙醇诱导的肝损伤进展时所必需。LPS信号出现于LPS和LPS结合蛋白(LBP)附着于细胞表面受体CD14时。CD14由LPS受体复合物的两个主要组分组成,能与Toll样受体4(TLR-4)相互作用。LPS受体复合物活化后可刺激多个信号转导级联反应。缺乏LPS受体组分(CD14、TLR-4、LBP)的小鼠,即使长期予乙醇灌胃亦不发生肝损伤。
TLR-4信号可由MyD88依赖性和非依赖性途径介导。MyD-88依赖性信号以LPS刺激后迅速出现TNF-α表达为特征,MyD88非依赖性信号的激活则较为缓慢,可使Ⅰ型干扰素(IFN)和IFN依赖性基因表达增加。长期乙醇暴露可使Kupffer细胞对TLR-4-MyD88依赖性反应,如MAPK、NF-κB快速激活和TNF-α表达致敏。在多个长期乙醇暴露模型中,大鼠和人类血TNF-α水平均增高。TNF-α受体Ⅰ(TNFR1)是对TNF-α信号产生应答的表面受体,TNFR1缺陷小鼠和静脉注射抗小鼠TNF-α多克隆抗体兔血清的小鼠,暴露于乙醇后不出现肝损伤征象。近期资料提示,TLR-4信号的MyD88非依赖性/TRIF(Toll-interleukin-1 receptor domaincontaining adapter IFN-β)依赖性通路亦为长期乙醇暴露诱导肝损伤的关键成因。然而,目前关于长期乙醇暴露对MyD88非依赖性信号调节的影响所知甚少。综合上述研究结果,可以认为LPS-TLR-4信号和Kupffer细胞活化对乙醇诱导的肝损伤的进展均具有关键作用。
HSC的活化:活化的HSC是受损肝脏中胶原表达增加的主要来源,亦为肝纤维化的标志。肝纤维化或称瘢痕化,以ECM蛋白合成增加和降解减少所致的ECM过度沉积为特征。
正常肝脏中的HSC具有脂肪细胞样表型,胞质中含有富含维生素A的脂滴。初始静息型HSC对肝损伤产生应答,形成信号;随后这些信号诱导HSC从静息表型转分化为活性表型。促纤维化信号如血小板源性生长因子刺激HSC增殖并移行至损伤区域。此外,自分泌和旁分泌信号如转化生长因子-β(TGF-β)和结缔组织生长因子亦能刺激HSC活化和纤维形成。活化HSC的纤维化表型以Ⅰ型胶原过度合成和产生收缩能力为特征。在起始信号活性持续,以及由HSC和肝内其他类型细胞产生的多种自分泌和旁分泌介质的共同作用下,HSC维持于活化状态。
肝损伤时HSC与血管生成的相互作用
无论是在肝脏发育时还是肝损伤时,HSC均能促成肝脏血管生成,在血管生成时起肝脏特异性毛细血管周细胞的作用。然而在肝损伤时,此作用可能与典型周细胞有一定差异。这一差异与两种不同类型的微血管系统(肝窦和稍大一些的血管)一起,成为肝脏血管生成的独有特征。HSC在肝脏再生时血管生成中的作用已得到充分阐述,关于HSC介导的血管生成在肝损伤纤维化形成时起关键作用的研究亦日益增多。肝脏修复时,血管生成异常伴肝窦重塑受损,导致肝窦内皮细胞毛细血管化和肝窦血管间发生分流。要形成具有功能的血管,需对细胞和分子网络进行密切调节,血管内皮生长因子(VEGF)、TGF-β、肝细胞生长因子等细胞因子对血管生成具有重要调节作用。HSC分泌的血管生成素-1是一种血管源性细胞因子,为血管重塑所必需。抑制血管生成素-1信号可改善长期CCl4暴露或胆管结扎引起的肝纤维化。
ALD发生过程中实质细胞与非实质细胞间的相互作用
肝纤维化的病理生理学机制涉及肝脏中不同类型细胞间复杂的相互作用。肝脏先天性免疫系统的细胞组分亦为各种损伤致肝纤维化发生的常见要素。Kupffer细胞耗竭可减轻常用的CCl4肝损伤模型的肝纤维化。浸润的嗜中性粒细胞、NK-T细胞、T细胞、B细胞以及肝细胞、肝窦和血管内皮细胞共同促成纤维化形成,NK细胞则可抑制肝纤维化。
促炎细胞因子的产生是损伤致纤维化形成中的关键步骤。其他介质还包括ROS、活性氮簇和凋亡细胞小体。这些介质均可在乙醇暴露时被激活。事实上,越来越多的研究表明,许多在乙醇诱导的肝脏炎症反应中起关键作用的因素亦为肝纤维化所必需,如乙醇诱导肝脏脂肪变性和炎症反应时,均需有LPS受体TLR-4表达。近期研究亦证实了TLR-4信号对HSC的作用,及其在肝纤维化形成中的作用。最近资料表明,乙醇亦能抑制肝脏NK细胞功能,减弱其抗纤维化作用。
结语
长期酗酒是肝病最常见的病因之一,且可与其他环境和(或)遗传因素相互作用,加速肝纤维化和肝损伤的进展,如酒精摄入可加速人类丙型肝炎和动物CCl4模型的肝纤维化。乙醇致肝纤维化的机制,及其与其他肝毒素协同作用加速纤维化的能力,均未完全阐明。乙醇的促纤维化作用可能或至少部分与乙醇损伤肝脏,使肝细胞对损伤的易感性增强有关。其他分子机制亦可能参与了乙醇加速肝纤维化的作用,如乙醇可诱导LPS产生增加,通过与Kupffer细胞上的TLR-4和HSC相互作用,刺激炎症反应,激活HSC。乙醇诱导的氧化应激、高反应性醛类的产生和(或)细胞凋亡亦可能加速肝纤维化。进一步了解乙醇对肝纤维化发病机制的复杂影响,将有助于ALD患者治疗和干预措施的开发。
(略)
译自Cohen JI,Nagy LE.Pathogenesis of alcoholic liver disease: interactions between parenchymal and nonparenchymal cells.J Dig Dis,2011,12(1):3-9.
