HPLC-MS/MS测定绿豆中黄曲霉毒素G2、G1、B2、B1
2011-05-01王少敏
许 勇,王少敏,毛 丹,郑 荣,王 柯,季 申
(上海市食品药品检验所,上海 201203)
黄曲霉毒素污染问题,已经成为世界各国密切关注的问题之一。黄曲霉毒素是一组具有强毒性和致癌性的次级真菌代谢产物,全球性污染粮食及其制品,给人类的健康造成严重威胁[1]。在黄曲霉产生的十余种毒素中,黄曲霉毒素G2、G1、B2、B1的毒性及致癌性最强。中药材或中成药中黄曲霉毒素的分析方法的研究近年来发展十分迅速,已有研究表明中药材及中成药中黄曲霉毒素的污染不可忽视[2~5]。为了保证临床用药的安全性,本文建立了用高效液相色谱串联三重四极杆质谱对中药绿豆中的黄曲霉毒素G2、G1、B2、B1进行了测定。方法简便、准确、专属性强。
1 仪器与试药
1.1 仪器 液相色谱串联质谱仪:美国Agilent公司1200液相色谱-API5500三重四极杆串联质谱仪,配有电喷雾离子源(ESI);多功能真空样品处理器;氮气吹干仪(N-EVAP 112型,美国 Organomation Associates,Inc公司);分析天平(BS2202/TE-612-L/CP225D型电子天平,德国Sartorius公司);HLB 柱(3mL/60mg,Waters,095A)。
1.2 试药 黄曲霉毒素混合对照品溶液(美国SUPELCO公司提供 Lot:LB 33367,黄曲霉毒素 G2、G1、B2、B1的浓度分别为 0.3 μg·mL-1、1.0 μg·mL-1、0.3 μg·mL-1、1.0 μg·mL-1);绿豆(批号:090721,产地:浙江)。甲醇、乙腈均为色谱纯(MERCK公司),试剂均为分析纯[中国医药(集团)上海化学试剂公司]。
2 方法与结果
2.1 液相色谱条件 色谱柱:Phenomenex SB-C18(2.0mm×50mm,4μm)。以 10mmol·L-1醋酸铵溶液为流动相 A,以甲醇为流动相B,按表1进行梯度洗脱;体积流量:1.0mL·min-1;柱温:25 ℃;进样量10μL。
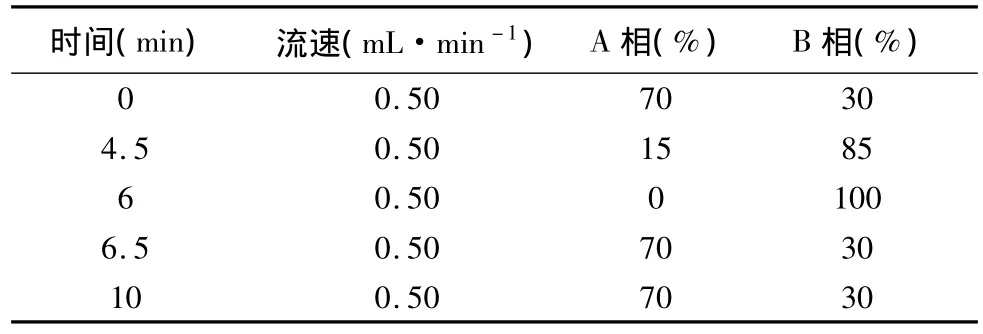
表1 流动相条件
2.2 质谱条件 经过对毛细管出口电压、碰撞池能量等质谱参数的优化,最终确定黄曲霉毒素G2、G1、B2、B1质谱条件 见表2。
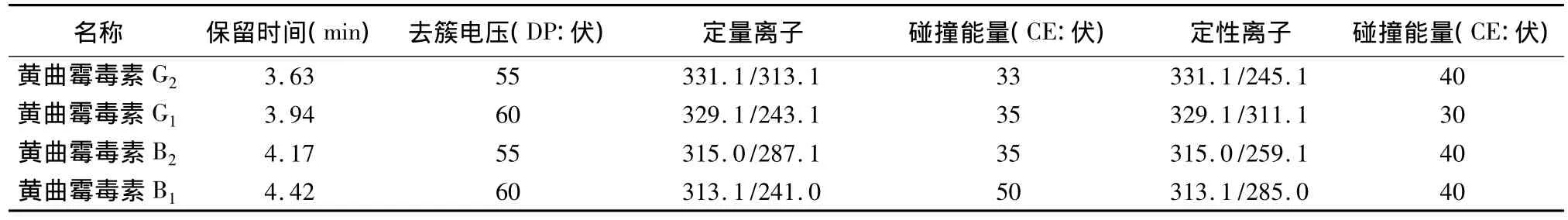
表2 黄曲霉毒素G2、G1、B2、B1检测的质谱参数
2.3 溶液的配制
2.3.1 对照品溶液的制备 取黄曲霉毒素混合对照品溶液,加50%甲醇制成含黄曲霉毒素G2、G1、B2、B1浓度分别为 30 ng·mL-1、100 ng·mL-1、30 ng·mL-1、100 ng·mL-1的对照品溶液。
2.3.2 供试品溶液的制备 取样品粉末(过二号筛)5 g,精密称定,精密加入70%甲醇50mL,超声处理30min,离心5min(离心速度2500 r·min-1),精密吸取上清液 10mL,用水稀释至20mL,摇匀,取10mL,通过已经处理好的HLB(先用甲醇2mL洗脱,再用水2mL洗脱)柱(流速3mL·min-1),随后用 30%甲醇2mL 洗脱(流速6mL·min-1),弃去洗脱液,再用1.5mL甲醇洗脱(流速1mL·min-1),收集甲醇洗脱液,用水稀释至2mL,摇匀,即得。
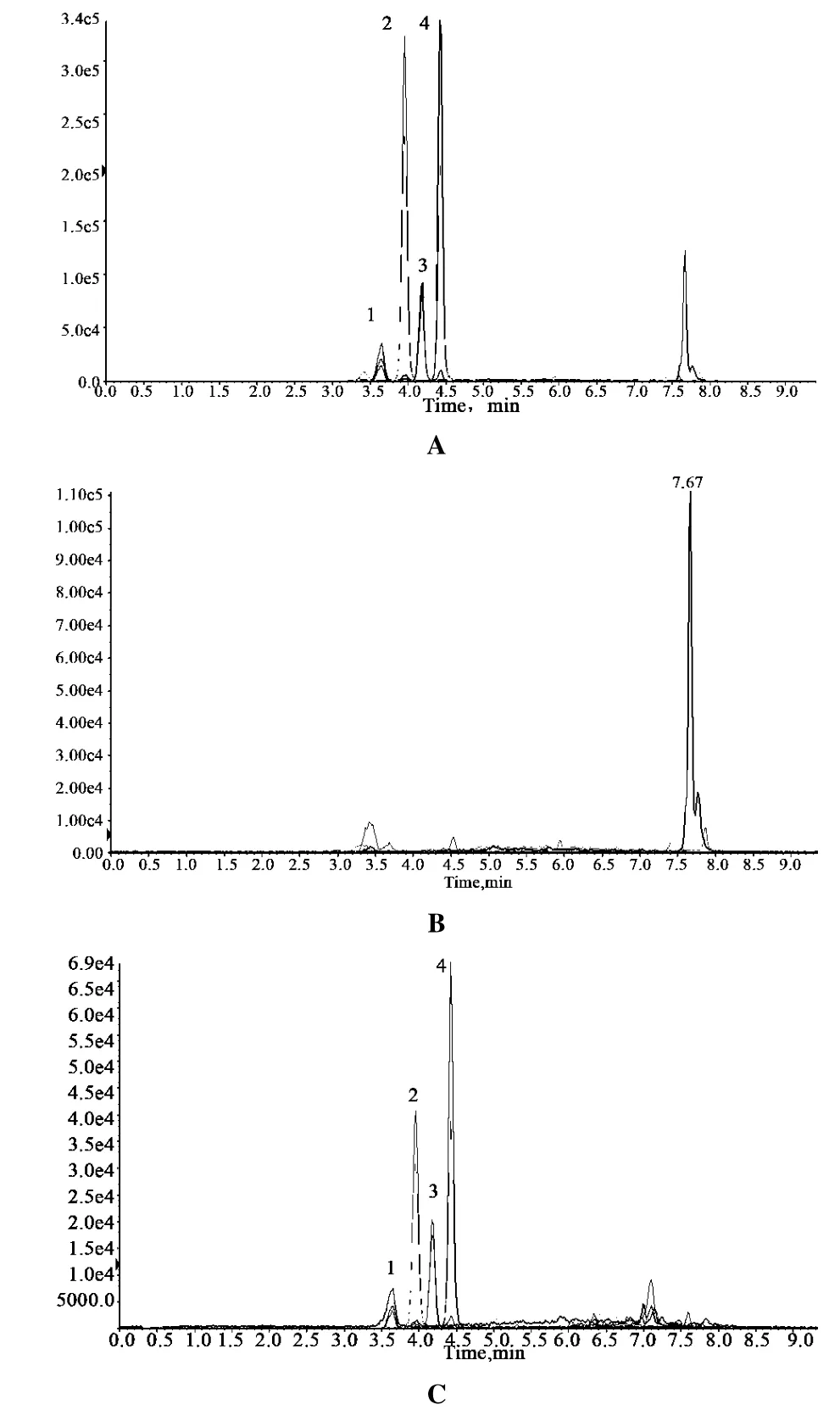
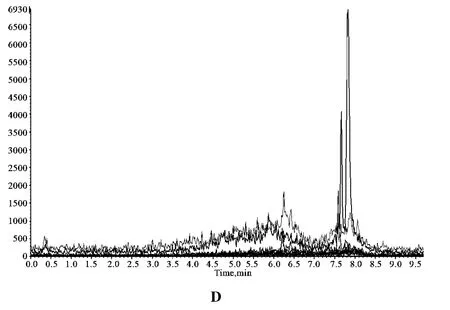
图1 黄曲霉毒素G2、G1、B2、B1对照品(A)、绿豆样品(B)、绿豆加入对照品(C)、阴性对照(D)的总离子流色谱图
2.3.3 阴性对照溶液的制备 取缺绿豆的样品按供试品溶液的制备方法制备阴性对照溶液。阴性样品溶液在与黄曲霉毒素G2、G1、B2、B1色谱峰相同位置处无干扰峰,表明样品中其他成分对黄曲霉毒素G2、G1、B2、B1的测定无干扰,见图1。
2.4 方法学考察
2.4.1 线性关系考察 取黄曲霉毒素G2、G1、B2、B1对照品溶液,加75%甲醇配制成5个不同浓度的系列对照品溶液。取样品5 g,一式5份,按正文所述方法制备供试品溶液,将供试品溶液于40℃氮吹至干,分别精密加入上述系列对照品溶液2mL,涡旋混匀,作为基质混合对照品溶液。精密吸取各基质混合对照品溶液10μL,进样分析,记录各待测组分色谱峰面积,以各成分的进样量为横坐标(X),各成分的峰面积为纵坐标(Y),进行回归分析,校正曲线,结果见表3。

表3 回归方程和相关系数
2.4.2 检测限的测定 取基质混合对照品溶液逐步稀释后,进样,以峰高为基线噪音3倍计,黄曲霉毒素G2、G1、B2、B1的最低检测限分别为 0.11 μg·kg-1、0.03 μg·kg-1、0.01 μg·kg-1、0.02 μg·kg-1。
2.4.3 精密度试验 取基质对照品溶液(黄曲霉毒素G2、G1、B2、B1浓度分别为 3 ng·mL-1、10 ng·mL-1、3 ng·mL-1、10 ng·mL-1),连续进样 6 次,记录峰面积,结果黄曲霉毒素 G2、G1、B2、B1峰面积的 RSD 值在0.8% ~2.0%范围内,表明测定精密度良好。
2.4.4 重复性试验 因样品未检出黄曲霉毒素,故采用加样方法进行重复性试验。取本品粉末(过二号筛)5 g(未检出黄曲霉毒素),一式6份,精密称定,精密加入黄曲霉毒素对照品溶液0.3mL,按正文方法制备供试品溶液,进样分析,记录峰面积,计算回收率。结果见表4,表明测定重复性良好。
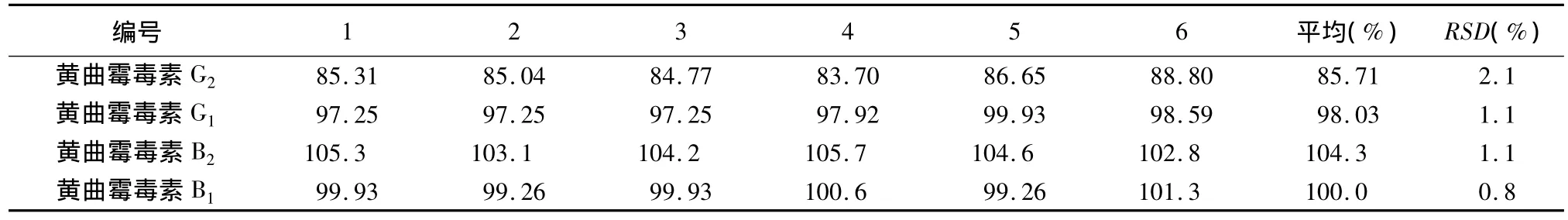
表4 重复性试验结果(%)
2.4.5 稳定性试验 取中间浓度回收率试验的供试品溶液,室温下放置 0、2、4、12、46 h,进样测定。计算黄曲霉毒素G2、G1、B2、B1浓度,其 RSD 值在 0.5% ~ 1.1% 范围内,表明供试品溶液在46 h内化学性质稳定。
2.4.6 回收率试验 取本品粉末(过二号筛)5 g(未检出黄曲霉毒素),一式9份,以3份为一组,分别精密加入黄曲霉毒素 G2、G1、B2、B1对照品溶液各 0.15、0.3、0.5mL,精密加入70%甲醇50mL,余同供试品溶液制备方法制得加样回收率供试品溶液。依法测定,计算回收率及相应RSD值,结果表明,本方法回收率试验结果良好,结果见表5~7。
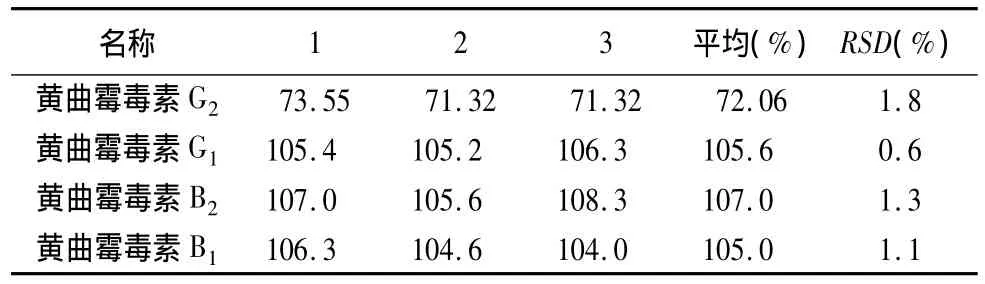
表5 低浓度添加水平回收率试验结果(%)(黄曲霉毒素 G2、G1、B2、B1 的添加浓度分别为 0.9 μg·kg-1、3 μg·kg-1、0.9 μg·kg-1、3 μg·kg-1)
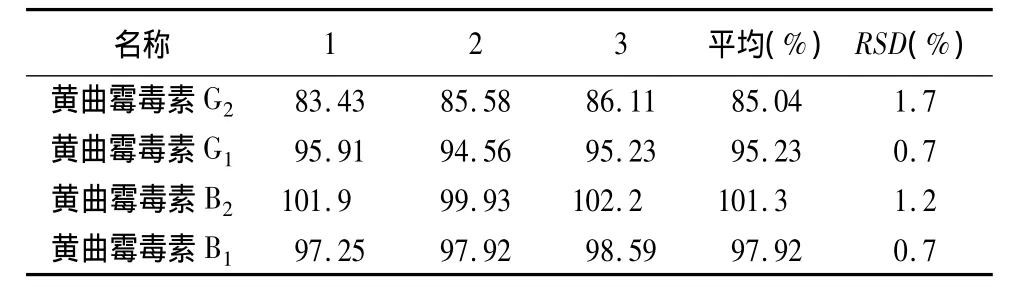
表6 中间浓度添加水平回收率试验结果(%)(黄曲霉毒素 G2、G1、B2、B1 的添加浓度分别为 1.8 μg·kg-1、6 μg·kg-1、1.8 μg·kg-1、6μg·kg-1)
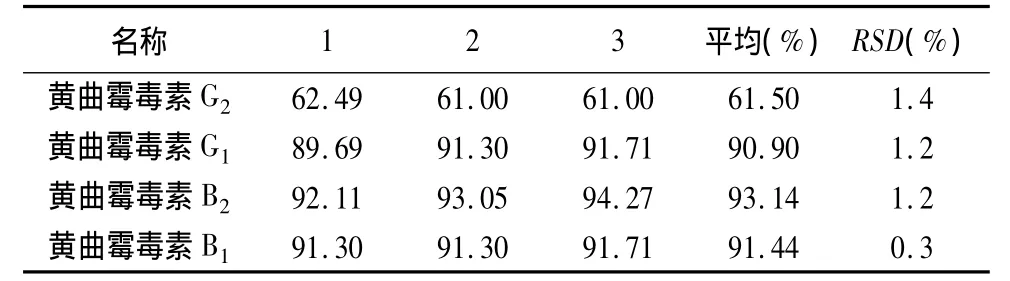
表7 高浓度添加水平回收率试验结果(%)(黄曲霉毒素 G2、G1、B2、B1 的添加浓度分别为 3 μg·kg-1、10 μg·kg-1、3 μg·kg-1、10 μg·kg-1)
2.4.7 样品测定 按拟订方法测定绿豆样品,结果未检出黄曲霉毒素 G2、G1、B2、B1。
3 讨论
3.1 基质效应的考察及其消除和补偿措施 基质效应指样品中除目标分析物以外的其他成分对待测物测定值的影响,即基质对分析方法准确测定分析物能力的干扰。根据基质对检测信号响应值的不同影响,可分为基质增强效应和减弱效应。增强效应即基质成分的存在减少了色谱系统活性位点与待测物分子作用的机会,使得待测物检测信号增强的现象;减弱效应是指基质成分的存在使仪器检测信号减弱的现象。不同的待测物由于化学结构与性质不同,在不同种类的基质中表现为不同的基质效应,本文在实验中对此进行了研究。
试验中,分别以混合对照品溶液和基质混合对照品溶液制备两条标准曲线,考察两条标准曲线对加样回收率结果的影响,结果发现,黄曲霉毒素 G2、G1、B2、B1存在基质减弱的现象。混合对照品溶液配制的标准曲线计算的回收率结果明显偏低。因此,为了保证测定结果准确可靠,本实验采用基质混合对照品溶液进行方法学的研究。
4 结论
目前,文献报道的中药中黄曲霉毒素的检测方法,多数为酶联免疫吸附法(ELISA)和免疫亲和柱荧光光度法。但是,因为中药的成分较为复杂,化学成分干扰较多,用ELISA试剂盒测定中药中黄曲霉毒素,假阳性率较高,不同厂家的试剂盒及各实验室测定结果差异较大。免疫亲和柱荧光光度法是2010年版《中国药典》附录中收载的中药中黄曲霉素G2、G1、B2、B1的通用测定方法。但此方法所用的免疫亲和柱价格比较昂贵,且有时也会出现假阳性。与上述方法相比较,本法采用HLB柱对样品进行净化后,通过液相色谱串联质谱对样品进行进样分析,即降低了分析的成本,提高了检测的灵敏度,又减少了假阳性,提高了测定的准确性,有较强的实用性。
[1]刘艳丽,程安春,汪铭书.黄曲霉毒素及其检测方法的研究进展[J].黑龙江畜牧兽医,2006,6:15 -17.
[2]任凤兰,马宏伟.应用试剂盒检测中药材及中成药黄曲霉毒素 B1[J].药物分析杂志,1997,17(4):280 -281.
[3]梁月秋,黄荣芳.中药污染黄曲霉毒素B1检测分析[J].中国现代应用药学杂志,2000,17(3):224 -226.
[4]Aziz NH,Youssef YA,El- Fouly MZ,et al.Contamination of some common medicinal plant samples and spices by fungi and their mycotoxins[J].Bot Bull Acad Sin,1998,39(4):279-285.
[5]Roy AK,Chourasia HK.Mycoflora,mycotoxin producibility and mycotoxins in traditional herbal drugs from India[J].J Gen Appl Microbiol,1990,36(5):295 -302.
