EPHB6受体型酪氨酸蛋白激酶在非小细胞肺癌中高甲基化失活
2011-04-26谷仲平陈宝莹第四军医大学基础部教学实验中心西安700唐都医院胸腔外科唐都医院放射科通讯作者mailyujunbyfmmueducn
铁 茹, 胡 浩, 谷仲平, 曲 萍, 于 军, 陈宝莹 (第四军医大学基础部教学实验中心, 西安 700;唐都医院胸腔外科;唐都医院放射科;通讯作者,E-mail:yujunby@fmmu.edu.cn)
肺癌是癌症患者的常见死因,而非小细胞肺癌(non-small cell lung cancer,NSCLC)是其中最常见的病理学类型[1,2]。远处转移是NSCLC引起死亡的最常见原因,在此过程中受体型酪氨酸激酶(receptor tyrosine kinase,RTK)发挥重要的作用[3,4]。促红细胞生成素产生肝细胞激酶(erythropoietin-producing hepatocyte kinase,EPH)受体是RTK家族中最大的亚家族,到目前为止,已在动物中发现有16个EPH 成员[5,6]。EPH 受体与 ephrin配体相互作用。EPH受体分为两类:EPHA(包括 EPHA1-10)和EPHB(包括EPHB1-6),这两类受体分别与各自的配体结合发挥作用。EPH受体与其配体结合之后产生一系列生物学效应,包括细胞间相互作用和细胞迁移[7,8]。EPHB6 作为 EPHB 受体之一,它的表达失活与人类多种肿瘤的发生和转移有关[9-15]。有文献报道,在神经母细胞瘤患者中,EPHB6的表达提示预后较好[9-11]。另外,在转移性黑色素瘤和具有转移潜能的浸润性乳腺癌细胞中,都发现EPHB6表达下降[13-15]。与正常肺组织相比,A549 NSCLC细胞系低水平表达 EPHB6[16],为此,我们选择A549细胞作为主要模型研究EPHB6表达失活的机制。
1 材料和方法
1.1 临床材料 收集2007-08~2010-07医院门诊正常肺组织患者9例,以及诊断为非小细胞肺癌(NSCLC)患者78例,其中未发生转移的NSCLC患者39例和发生转移的NSCLC 39例。
1.2 实验材料 Dulbecco’s Modified Eagle’s培养基(DMEM,Invitrogen公司,美国),10%的胎牛血清(FCS),2 mmol/L L-谷氨酸,100 ml青霉素、100 μg/ml链霉素,A549肺腺癌细胞,5-氮-2'-脱氧胞苷(Sigma,MO,USA),反转录试剂盒(Promega,Madison,Wisconsin,USA),限制性内切酶 Sma Ⅰ(Applied Biosystems,Foster City,CA,USA),DNAzol(Invitrogen,Carlsbad,CA,USA),EZ DNA Methylation-Gold TM Kit试剂盒(Zymo research Corp.,Orange,CA,USA),TOPO TA 克隆测序试剂盒(Invitrogen 公司),兔抗人 EPHB6(1 μg/ml,Santa Cruz,USA),RNA(Invit Promega,Madison,Wisconsin,USA rogen,Carlsbad,CA,USA),ERK(1∶1 000 稀释,Cell Signaling Technology,USA),小鼠抗人 β-actin(40 ng/ml,sigma,USA),化学发光法(ECL Plus,Amersham Pharmacia,Sweden)显色。
1.3 方法 实验分为三组:正常肺组织组(正常组)、未发生转移的NSCLC肿瘤组织组(对照组)、发生转移的NSCLC肿瘤组织组(病例组)。
1.3.1 细胞培养 A549肺腺癌细胞培养采用改良Dulbecco’s Modified Eagle’s培养基,含10%的胎牛血清(FCS),2 mmol/L L-谷氨酸,100 ml青霉素、100 μg/ml链霉素,于37℃、5%CO2的孵箱中培养。1.3.2 患者标本 主要的肿瘤标本和正常的肺组织取自NSCLC外科手术,将标本置于液氮中冷冻,保存于-80℃冰箱,采用组织学方法检查肿瘤标本的肿瘤细胞百分比,仅将含有70%肿瘤细胞的活体肿瘤组织用于后续的mRNA和蛋白质的表达分析及随后的DNA甲基化分析。为了排除个体间的差异对实验结果的影响,还比较了另外24例NSCLC患者的成对的肿瘤组织和正常肺组织中EPHB6的mRNA表达水平。
1.3.3 5-氮-2'-脱氧胞苷处理 A549细胞用由1 mmol纯水原液配置的100 μmol/L的5-氮-2'-脱氧胞苷处理。细胞每48 h补充一次新鲜培养液和5-氮-2'-脱氧胞苷,生长2-6 d。
1.3.4 RNA提取和逆转录 RIzol试剂分离总RNA,以1 μg RNA为模板,采用反转录试剂盒逆转录为cDNA。
1.3.5 实时定量RT-PCR测定基因表达水平 为了观察患者EPHB6的DNA甲基化水平,采用了甲基化敏感的SmaⅠ限制性内切酶进行研究。采用实时定量RT-PCR法对临床Ⅰ期和Ⅱ期NSCLC患者肿瘤组织的 EPHB6表达水平进行测定。用DNAzol(提取试剂)从每一个样本中提取1 μg基因组 DNA,等分为两份,每份0.5 μg,用甲基化敏感性限制性内切酶SmaⅠ消化,或者用限制性内切酶的缓冲液假消化作为对照。通过实时定量PCR用以下引物和探针对消化或假消化DNA进行分析:正向引物5'-CGGGCCCCCAGGATCTC,反向引物5'-TGCCACCCGCGTCTTCTC,探针 5'-FAM-CGGCGCCGAACGGACCCG-TAMRA。探针位于甲基化敏感的SmaⅠ位点。甲基化水平的计算公式如下:甲基化水平 =2ΔCT,ΔCT=CTT-CTN(T:处理的SmaⅠ;N:未处理的SmaⅠ)。
1.3.6 亚硫酸氢盐测序法检测启动子区域与CpG岛甲基化水平 为了研究NSCLC中EPHB6基因被抑制的机制,需要测定了EPHB6启动子的甲基化水平。选择了1例肿瘤组织中EPHB6表达水平较正常肺组织显著降低的NSCLC患者为研究对象,采用亚硫酸氢盐测序法检测EPHB6基因的启动子和第一个内含子区。用DNAzol提取基因组DNA,然后按照说明书用亚硫酸氢盐与EZ DNA Methylation-Gold TM Kit试剂盒处理。利用Methyl Primers Express version 1.0(Applied Biosystems,Foster City,CA,USA)设计的引物进行PCR。启动子区域的甲基化水平的分析基于以下引物:5'-GGGTTGCGTTCGTTTTAGTC-3'(正向)和5'-CGCCGAAAATCCCTAAAATT-3'(反向)。内含子区域的引物是5'-AGTTAGATTGGGGGTAGAGTAG-3'(正向)和5'-ATCTAACCAAACAACCACTTAA-3'(反向)。用TOPO TA克隆测序试剂盒克隆PCR产物。用至少9个带有正常或肿瘤组织的EPHB6启动子或内含子区的克隆用于筛选和测序。
1.3.7 Western blot检测 用冰冻的 PBS冲洗细胞,加入含150 mmol NaCl的冰冷RIPA缓冲液,冰上裂解30 min。20 000×g、4℃离心10 min内清除细胞裂解物。调整蛋白质浓度后,在SDS上样缓冲液中以95℃加热细胞裂解物10 min,进行聚丙烯酰胺凝胶电泳(PAGE,4%,Invitrogen公司)以分离蛋白,转到聚偏氟乙烯(PVDF)膜。蛋白检测:一抗,兔抗人EPHB6,兔抗人磷酸化或总ERK,小鼠抗人β-actin。采用辣根过氧化物酶(HRP)耦联二抗,化学发光法显色。
2 结果
2.1 NSCLC中EPHB6表达缺失 EPHB6表达水平的测定的结果见图1。与对照组相比,肿瘤组织中EPHB6 mRNA水平降低,最低者出现在手术治疗后发生转移的患者肿瘤组织中(P<0.05,见图1A)。另外24例NSCLC患者的肿瘤组织和正常肺组织患者中13例的EPHB6 mRNA表达水平降低(图1B)。另外,Western blot分析显示NSCLC肿瘤组织中的EPHB6蛋白水平降低(图1C)。

图1 EPHB6表达水平的测定Fig 1 Expression of EPHB6 in different groups
2.2 NSCLC中EPHB6高甲基化 实时定量RTPCR测定基因表达水平结果见图2所示。序列分析结果表明,EPHB6的启动子区富含CpG二联体(图2A),与正常肺组织相比,肿瘤样本的启动子区及第一内含子区有6处CpG岛的核苷酸甲基化水平的升高(图2B),而NSCLC患者的样本相对于正常肺组织显示了较高水平的甲基化(P<0.05,见图2C)。EPHB6基因甲基化水平升高的肿瘤样本EPHB6 mRNA的转录水平降低(r=-0.55,P<0.05,见图2D)。基因甲基化水平高的肿瘤组织EPHB6的转录水平低(P<0.05,见图2E)。
在mRNA水平和蛋白质水平上,A549细胞的EPHB6基因表达显著上升(图3A、B),而EPHB6甲基化程度下降(图3C)。
3 讨论
本研究综合运用基于甲基化敏感的SmaⅠ限制性内切酶的实时定量RT-PCR实验、亚硫酸氢盐测序实验和5-氮-2'-脱氧胞苷诱导基因重新表达等实验技术,发现在NSCLC中EPHB6基因启动子发生了较为广泛的甲基化。EPHB6基因启动子区的高甲基化与NSCLC中EPHB6 mRNA和蛋白质水平的下调密切相关。EPHB6表达缺失与NSCLC患者早期发生转移的风险升高相关。从而,首次阐明了NSCLC中EPHB6表达失活的机制。
文献报道乳腺癌中EPHB6启动子区域的CpG岛呈高甲基化状态[13]。另外,还发现EPHB6基因第一个内含子中的甲基化水平存在组织差异[17]。在SmaⅠ限制性内切酶处理后,对照组的肺组织酶切片段扩增水平显著下降,而肿瘤样本的酶切片段扩增水平不变,从而进一步证实了上述硫酸亚氢盐测序实验的结果。我们对14例NSCLC患者的研究发现,NSCLC样本相对于正常肺组织显示了较高水平的甲基化,而EPHB6基因甲基化水平升高的肿瘤样本EPHB6 mRNA转录水平降低的数据显示甲基化和低表达有显著的相关性,同时基因甲基化水平高的肿瘤组织EPHB6的转录水平降低。通过对经过去甲基化试剂5-氮-2'-脱氧胞苷处理的A549细胞的EPHB6的表达水平进行测定结果发现,在mRNA水平和蛋白质水平上,EPHB6基因的表达都显著上升,这种表达上升与EPHB6的甲基化程度下降有关。
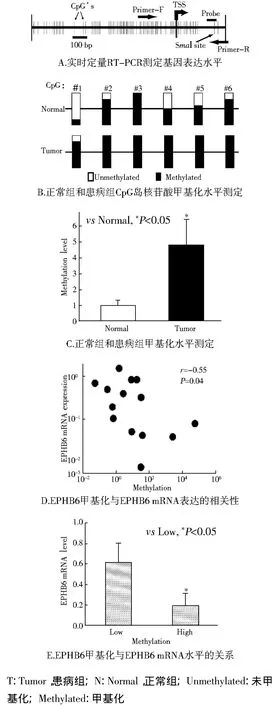
图2 实时定量RT-PCR测定基因表达水平Fig 2 Gene expression levels in NSCLC by RT-PCR
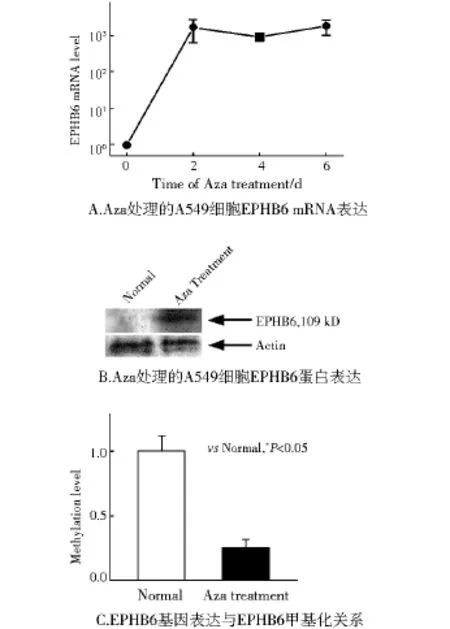
图3 Aza处理后EPHB6 mRNA水平及其甲基化的变化Fig 3 Changes of EPHB6 mRNA and methylation in NSCLC after Aza treatment
基因的活性受到遗传因素和表观遗传因素的双重调节。表观遗传(epigenetic)因素虽然不改变DNA序列,但可以通过对DNA的修饰而降低相关基因的转录活性[18]。表观遗传因素可以导致EPHB6“量”的变化,即EPHB6表达水平的降低会导致缺乏足够数量的分子来维持其正常的功能。目前已经明确,肿瘤的转移与多种癌基因的低甲基化过表达以及抑癌基因的高甲基化失活有关,甲基化导致基因沉默的频率高于突变、缺失等造成的基因沉默[19,20]。DNA序列中的 CpG 二联体是甲基化的靶点,DNA甲基转移酶(DNA methyl transferase,DNMT)以CpG二联体中的C为底物,将S腺苷甲硫氨酸(SAM)上的甲基转移到 C上,形成甲基化的mCpG。一些DNMT抑制剂被用于逆转抑癌基因的甲基化。CpG二联体以较大的密度分布于基因5'端,称为CpG岛。CpG岛一般为0.5-2 kb长,通常位于基因的5'端启动子区,也可延伸至外显子区,CpG岛的高甲基化将抑制该基因转录[23]。文献报道在多种人类肿瘤中 EPHB6 表达均被抑制[13,15,22],而EPHB6在乳腺癌细胞系中的转录失活是启动子区DNA高甲基化造成的[15],从而表明EPHB6的高甲基化沉默可能在多种类型肿瘤当中都会发生。
综上所述,NSCLC中EPHB6受体型酪氨酸蛋白激酶因启动子区高甲基化而沉默,EPHB6的失活与NSCLC的转移密切相关,提示通过降低EPHB6启动子DNA的甲基化水平激活其表达,有可能为预防NSCLC发生转移提供新的方法。
[1] Jemal A,Siegel R,Ward E.Cancer Statistics[J].CA Cancer J Clin,2009,59:225-249.
[2] van Zandwijk N,Mooi WJ,Rodenhuis S.Prognostic factors in NSCLC.Recent experiences[J].Lung Cancer,1995,12(Suppl 1):S27-33.
[3] Ansari J,Palmer DH,Rea DW,et al.Role of tyrosine kinase inhibitors in lung cancer[J].Anticancer Agents Med Chem,2009,9:569-575.
[4] Santos ES,Blaya M,Raez LE.Gene expression profiling and nonsmall-cell lung cancer:where are we now[J].Clin Lung Cancer,2009,10:168-173.
[5] Menzel P,Valencia F,Godement P,et al.Ephrin-A6,a new ligand for EphA receptors in the developing visual system[J].Dev Biol,2001,230:74-88.
[6] Eph Nomenclature Committee.Unified nomenclature for Eph family receptors and their ligands,the ephrins[J].Cell,1997,90:403-404.
[7] Castaño J,Davalos V,Schwartz S Jr,et al.EPH receptors in cancer[J].Histol Histopathol,2008,23:1011-1023.
[8] Campbell TN,Robbins SM.The Eph receptor/ephrin system:an emerging player in the invasion game[J].Curr Issues Mol Biol,2008,10:61-66.
[9] Tang XX,Evans AE,Zhao H,et al.Association among EPHB2,TrkA,and MYCN expression in low-stage neuroblastomas[J].Med Pediatr Oncol,2001,36:80-82.
[10] Tang XX,Evans AE,Zhao H,et al.High-level expression of EPHB6,EFNB2,and EFNB3 is associated with low tumor stage and high TrkA expression in human neuroblastomas[J].Clin Cancer Res,1999,5:1491-1496.
[11] Tang XX,Zhao H,Robinson ME,et al.Prognostic significance of EPHB6,EFNB2,and EFNB3 expressions in neuroblastoma[J].Med Pediatr Oncol,2000,35:656-658.
[12] Fox BP,Kandpal RP.Invasiveness of breast carcinoma cells and transcript profile:Eph receptors and ephrin ligands as molecular markers of potential diagnostic and prognostic application[J].Biochem Biophys Res Commun,2004,318:882-892.
[13] Fox BP,Kandpal RP.Transcriptional silencing of EphB6 receptor tyrosine kinase in invasive breast carcinoma cells and detection of methylated promoter by methylation specific PCR[J].Biochem Biophys Res Commun,2006,340:268-276.
[14] Hafner C,Bataille F,Meyer S,et al.Loss of EphB6 expression in metastatic melanoma[J].Int J Oncol,2003,23:1553-1559.
[15] Hafner C,Schmitz G,Meyer S,et al.Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers[J].Clin Chem,2004,50:490-499.
[16] Mueller WC,von Deimling A.Gene regulation by methylation[J].Recent Results Cancer Res,2009,171:217-239.
[17] Irizarry RA,Ladd-Acosta C,Wen B,et al.The human colon cancer methylome shows similar hypo-and hypermethylation at conserved tissue-specific CpG island shores[J].Nat Genet,2009,41:178-186.
[18] Yoo CB,Jones PA.Epigenetic therapy of cancer:past,present and future[J].Nat Rev Drug Discov,2006,5:37-50.
[19] Ehrlich M.Cancer-linked DNA hypomethylation and its relationship to hypermethylation[J].Curr Top Microbiol Immunol,2006,310:251-274.
[20] Baylin SB,Schuebel KE.Genomic biology:the epigenomic era opens[J].Nature,2007,448(7153):548-549.
[21] Herman JG,Baylin SB.Gene silencing in cancer in association with promoter hypermethylation[J].N Engl J Med,2003,349:2042-2054.
[22] Tang XX,Robinson ME,Riceberg JS,et al.Favorable neuroblastoma genes and molecular therapeutics of neuroblastoma[J].Clin Cancer Res,2004,10:5837-5844.
