莽草酸的合成研究现状
2011-02-20寇玉辉
寇玉辉, 雷 叶
1.西陇化工股份有限公司,广东 汕头 515064;2.旭丽电子广州有限公司, 广东 广州 510663)
0 引 言
莽草酸是一种重要的生物活性物质,在植物和微生物代谢中,它是许多天然产物包括芳香族氨基酸、生物碱、酚类等物质的生物合成中间体,即莽草酸途径[1].莽草酸也是一些特殊酶的抑制剂,是合成神经氨酸酶抑制物的一种重要原料,可用来合成抗流感药物[2].
早在1885年,莽草酸就被发现并分离得到,20世纪50年代出现了莽草酸的化学合成方法.生物提取法虽然也得到了发展,但成本较高,与此同时还出现了微生物发酵法.自2003年禽流感侵袭以来,莽草酸是有效对付致命性H5H1禽流感病毒药物“达菲”的重要成分,因此莽草酸的需求日益增大.目前,八角茴香是工业上莽草酸的主要来源,但生产成本很高,因此开发莽草酸新型合成法具有重要意义.
1 莽草酸结构及分布
莽草酸(Shikimic acid,SA)属酚类化合物,化学名为[3R-(3α,4α,5β)]-3,4,5- 三羟基-1- 环己烯-1-羧酸,分子式为C7H10O5,结构见图1,具有手性,其中1a具有开发药物的功效.它是1885年从日本莽草“Shikimic-Ki(Illiciumanisatum)”分离提取而得名.到目前为止,莽草酸除了在八角茴香中(含量达12.57%)被发现大量存在外,在番茄叶、醋栗果实、金丝桃等160多种植物中其存在也被发现.

图1 莽草酸结构
2 莽草酸的化学合成
莽草酸是合成很多药物的原料,其立体专一性历来为人们关注,早在1959年Smissman等[3]就立体专一地合成出dl-莽草酸.1968年,有科研人员对D(-)莽草酸全合成中的中间体做了详细的描述[4].1971年,又报道了以D-树胶醛糖为原料合成出立体专一、具有光学纯度的莽草酸[5].
早期方法虽有立体选择性好、中间某些合成步骤反应条件简单、产率高等优点,但是大都步骤较多,总收率不高.近年来合成方法有所改进,取得了一些进展.
2.1 Diels-Alder合成法
Takashi等[6]以人造纤维2为初始原料,经过17步反应得到了(-)-莽草酸7,反应示意图如图2.
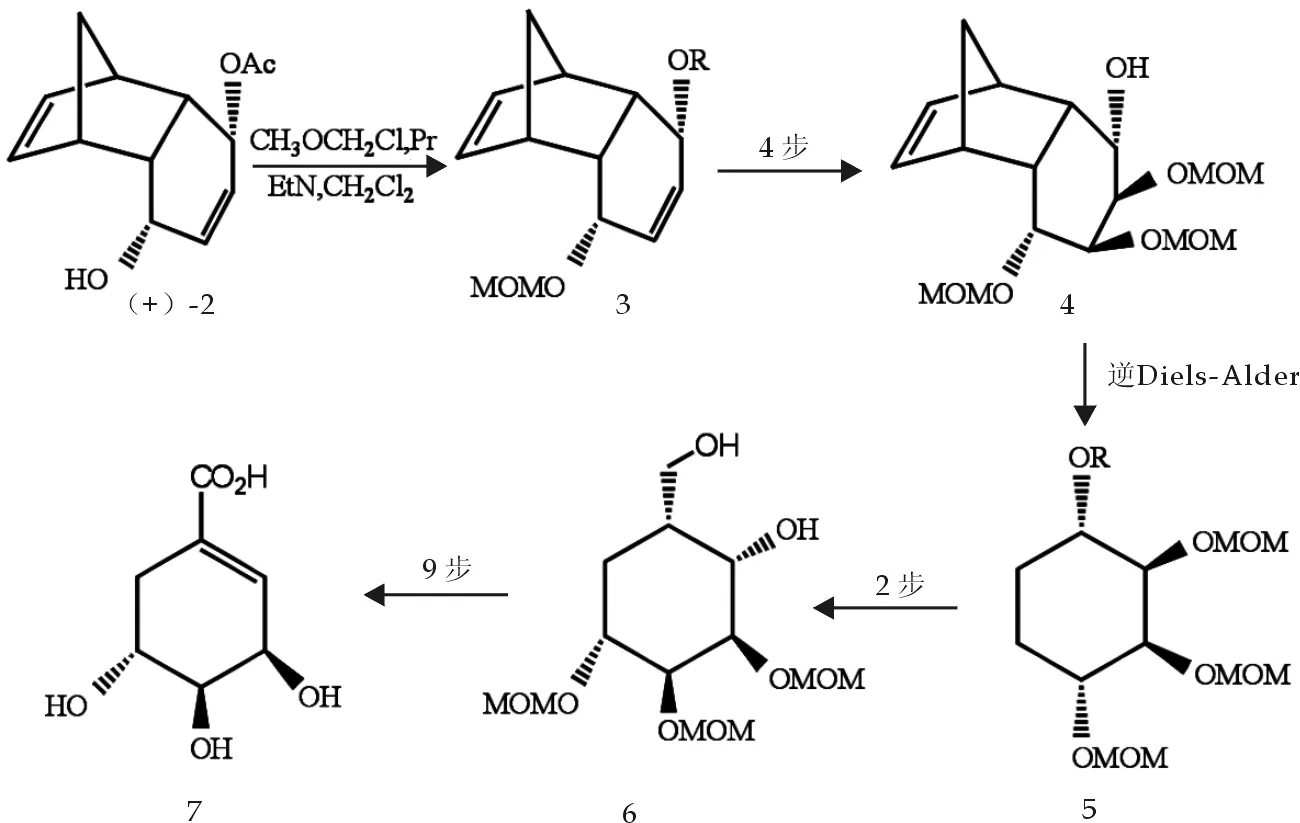
图2 莽草酸合成反应式
此反应一开始就用氯甲醚(MOMOCl)保护羟基,一直到倒数第二步才还原,这是很好的思路,但反应步骤太多,总收率也很低.
KouHiroya等[7]以纯的(-)-(1S,4R,4aS,5S,8aS)-8-(二甲叔丁基硅氧基)-4,4a,6,7,8,8a-六氢-1,4- 二甲基萘-5(1H)-酮7做为起始反应物,经13步制得莽草酸,收率达到40%,其中9-10为逆Diels-Alder反应,产率近100%,对我们以后进行此类反应有很大启发,步骤如图3.
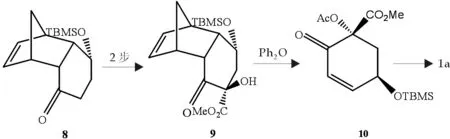
图3 莽草酸合成反应式
Naoyuki等[8]以(+)-(1S,4R,4aR,5R,8S,8Ar)-1,5,8,8a-四氢-5,8,-二羟氢-1,4-亚甲基萘-4a(4H)-酸羧酸甲酯11为起始反应物,经11步反应制得莽草酸,步骤如图4.
虽然步骤少、反应条件也较简单,但其中逆Diels-Alder反应产率仅为52%,总产率也仅有14.8%.
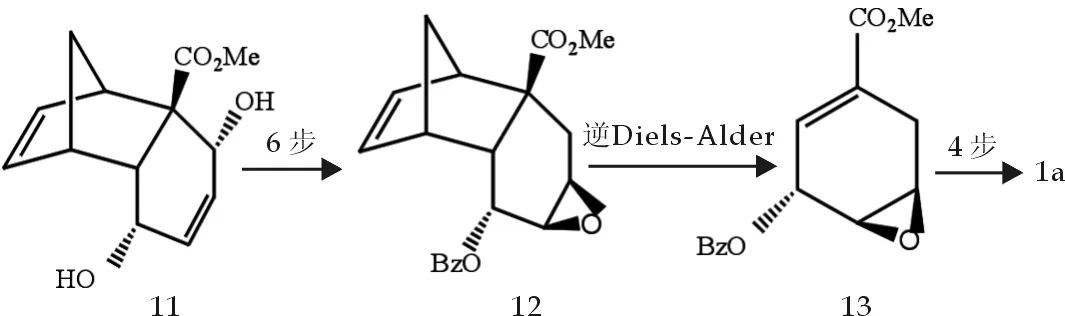
图4 莽草酸合成反应式
1997年,Javier等[9]以亚硫酰丙烯酸酯14为原料,通过Mannichi反应,经过中间体15,得到二烯亲和体16,在高压下与呋喃发生Diels-Alder反应得到4种混合物17、18、19和20,其中18产率为59%,消去SOTOl得到环氧冰片21,再通过与LHMDS反应断裂氧桥键得到环己烯22,通过加上保护基制备出它的TBDMS衍生物23,再除去保护基得到24,反应式如图5.
此方法合成出(+)-莽草酸,且较为简易,但其中Diels-Alder反应产生4种混合物,分离困难且产率不高,还有其中一步产率仅为28%,如果能够提高Diels-Alder反应的选择性和提高个别低产率反应的产率,此方法将大有可为.
Surachai等[10]用顺丁烯二酐25为原料,通过乙二烯26的Diels-Alder反应得到主加成产物27,再通过与28反应得到产物29(R=OAC),收率为84%.酐29通过甲醇分解作用得到30(收率83%),再通过甲基化氢解作用得到酸31,通过Hundiecker反应转变为62∶38的混合物32,33,后再转化为莽草酸酯34,其收率为53%,34通过酸解得到D-葡萄糖和4-epi-莽草酸,通过离子交换柱分离后,得到莽草酸,产率80%,反应式如图6.
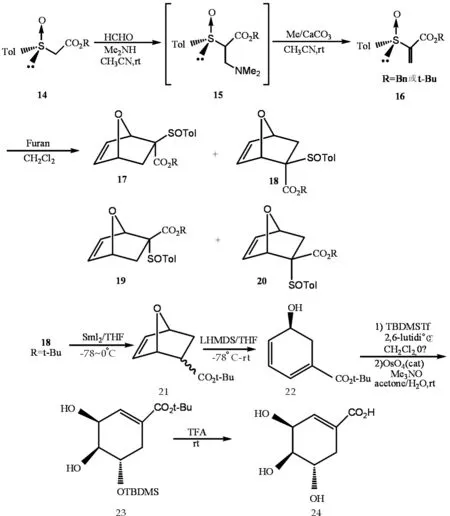
图5 莽草酸合成反应式
虽然此法步骤仍然较多,产率低,且操作麻烦,工业上应用价值不大,但它首次成功地合成了莽草酸基配糖体并首次完成了对4-epi-莽草酸的不对称合成,对以后的相关合成有重大的指导意义.
2.2 碳水化合物转化法
George等[11]用D-甘露糖为原料酯化得莱苏糖35,烷基化作用后得磷酸酯36,再在一定条件下转化为亚甲基莽草酸酯37,又经过脱酰作用得38,在碱性条件下水解得到(3R,4S,5R)- 莽草酸.此方法原料易得,且反应步骤较少,但是总产率仍较低,仅为25%,反应式如图7.
Shen等[12]以D-核糖39为原料经12步合成分别得到(-)-5- epi-莽草酸40和(-)-5- epi-莽草酸酯41,虽原料简单易得,但过程中的副产物较多,分离过程复杂,产率低,40为16.4%,41为12.7%,不适于工业化生产,反应式如图8.
2.3 以奎宁为原料合成
1999年,日本专利[13]报道了以奎宁酸为原料制得莽草酸前体而得到莽草酸的方法.由奎宁酸42酯化得到酯43,再通过水解和烷基化得到44,又经脱水反应得莽草酸前体45和46,45通过脱保护作用得莽草酸前体47,最后经过脱保护得(-)-莽草酸,反应式如图9所示.
此反应过程在由44制45的关键步骤中,前体45的产率可高达80%以上,且重结晶纯化后可以回收99%.此法使得对莽草酸前体的合成能做到高产率、高质量、高纯度且低耗费.这对莽草酸的工业化合成有重要指导意义.
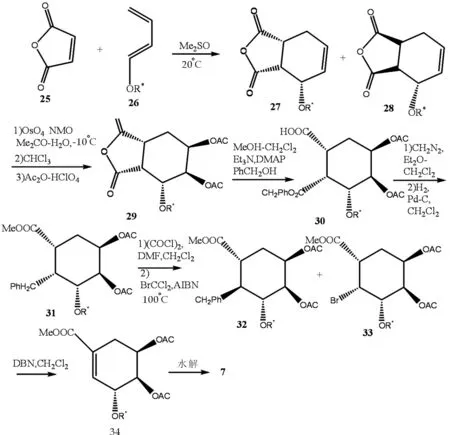
图6 莽草酸合成反应式

图7 莽草酸合成反应式
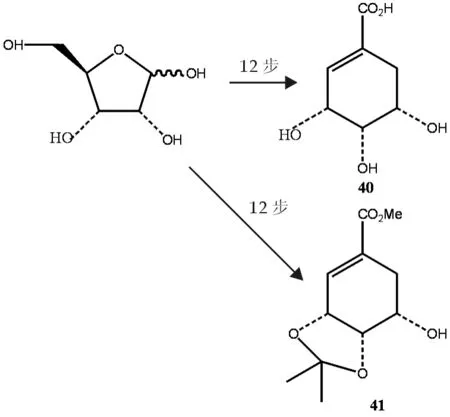
图8 莽草酸合成反应式
2.4 其它方法
Johnl等[14]报道了以合成分支酸过程中的中间产物48为原料而合成(-)-莽草酸的方法.48水解后得到前体49,再水解后得(-)-莽草酸,反应式如图10所示.
此反应过程各步产率都很高,且反应条件简便易得,总分离收率可高达94%.但49毕竟只是其它反应的一个中间产物,如果能提高它的产率或能找到更好的合成、分离或生物提取49的方法,将会大大简化(-)-莽草酸的合成工艺.
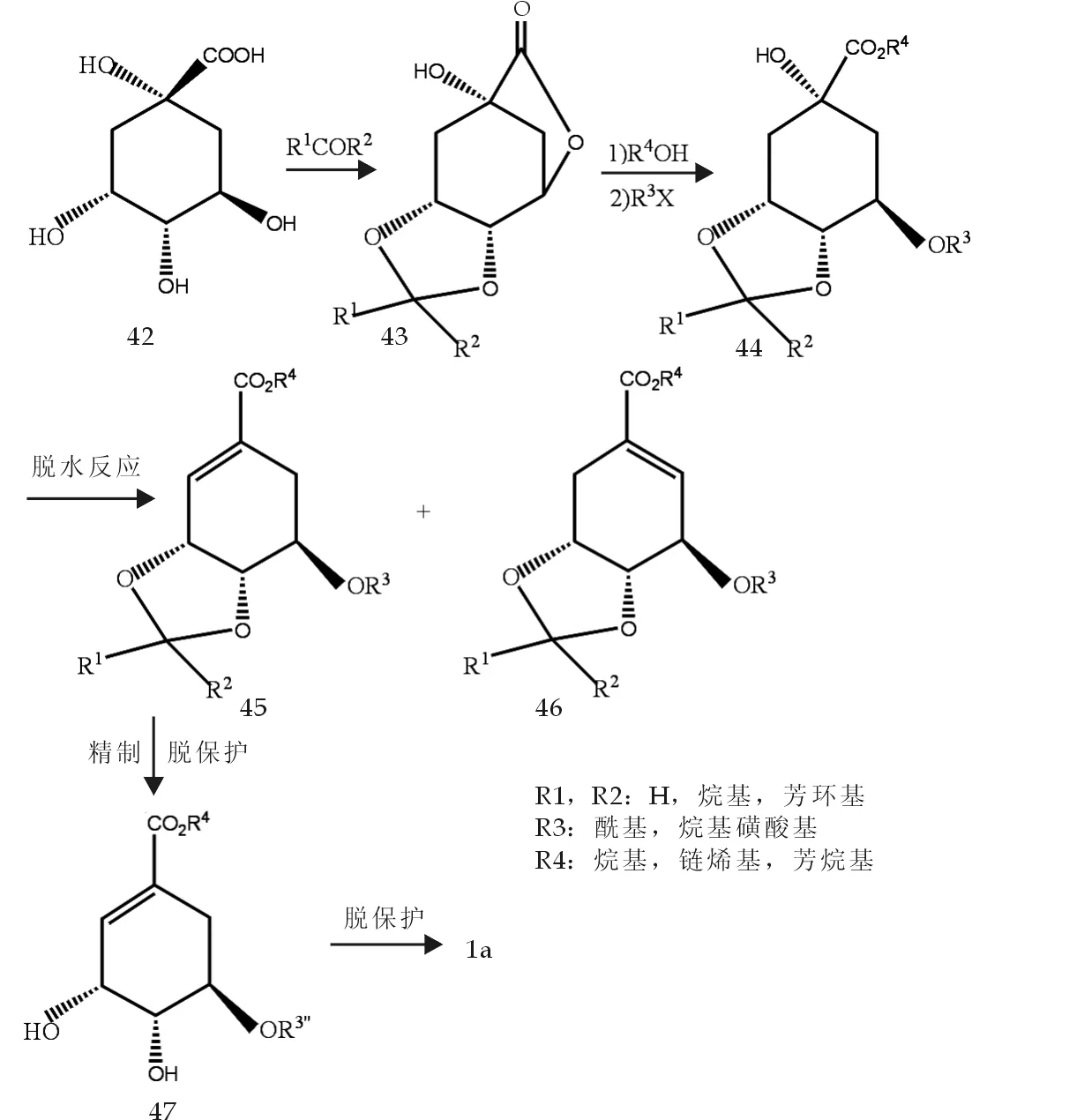
图9 莽草酸合成反应式
3 微生物(酶)法合成
1979年Ryoyasu等[15]最初报道从茶树中提取了与莽草酸的合成有关的4种酶:3-deoxy-D-arabino-heptulosonic acid-7-磷酸合成酶(EC.4.1.2.15)、DHQ合成酶、DHQase(EC.4.2.1.10)、SORase(EC.1.1.1.25),并对其性质进行了阐述,此后,4种酶在莽草酸合成途径中被大量利用.
根据同位素示踪研究和发酵条件研究结果,Chandrand等人[16]以葡萄糖为基质,采用E.coliSP1.1pts/ pSC 6.090 B 菌在1 L发酵罐中分批发酵合成莽草酸后,扩大至10 L规模并添加酵母浸液,发酵产物莽草酸浓度达87 g/L,产率为36%,但这与理论计量产率86%相差甚远,其生物合成途径如图11所示.葡萄糖经糖酵解成磷酸烯醇式丙酮酸(PEP)和4-磷酸赤藓糖(4EP),在DAHP合成酶的催化下,生成DHQ,然后经一系列酶促反应合成3-脱氢莽草酸,最后在脱氢酶的作用下得到莽草酸.在整个合成过程中,PEP对DAHP合成酶起反馈抑制作用,是莽草酸合成途径的一个重要控制因素,提高PEP的转化率就会导致奎尼酸和3-脱氢奎尼酸等副产物量增多,因此有必要通过改变发酵条件来提高产率.
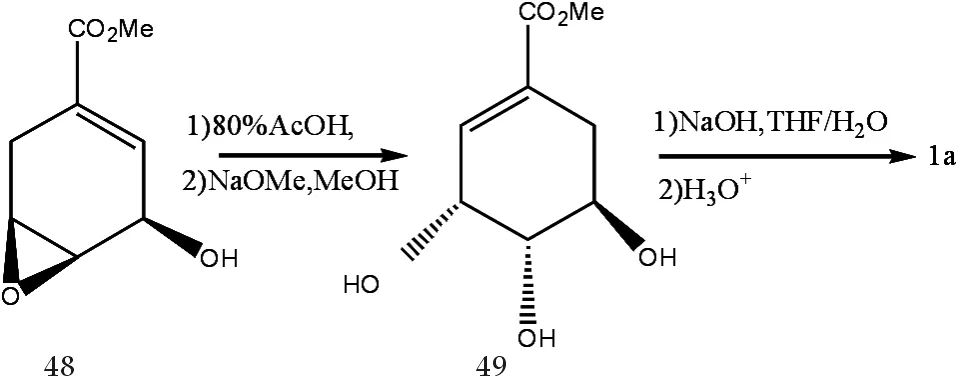
图10 莽草酸合成反应式
通过基因调控手段筛选基因工程菌,优化合成条件可以有效提高产量.Louise Johansson等人[17]利用上述合成途径,通过比较菌株(W3310)与莽草酸激酶Ⅱ缺失型菌株Escherichiacoli(W3310.shik1)在磷酸盐受限和碳源限制的条件下合成莽草酸的量,结果显示采用W3310.shikl在磷酸受限条件下产率更高,是碳源受限条件下的两倍,达(0.059±0.012 VS. 0.024±0.005 C-mol/ C-mol),副产物也最低,但是细胞溶解导致发酵液下游处理困难.Marco Kramer等人[18]也对上述合成代谢途径进行了综述,分别讨论了碳源的吸收、调节途径、中间代谢和涉及莽草酸摄入的芳香族途径,对提高产物产率和减少副产物有重要影响.
4 结束语
莽草酸除了可由天然原料直接分离得到,还有多种化学合成与生物合成法.目前工业上只能从八角茴香中提取大量的莽草酸以满足市场的需求,但是每30 kg八角茴香只能加工出1 kg莽草酸,化学合成的发展则可以满足大规模工业化的要求.以微生物细胞和酶为代表的生物催化剂,已成为现代化学合成中不可或缺的有利工具,需要配合我国制药工业发展的具体情况研究相关产品.普通环境中的有机合成有时只能得到无旋光的外消旋混合物,而用微生物法则易得到具有旋光活性的相对较纯的产物,也具有很高的实用价值.天然莽草酸的主要结构区别在于C3、C4和C5位的侧链,酶法可以保持起特殊作用的结构和构型.寻求更好更廉价的方法合成莽草酸有很好的应用前景.
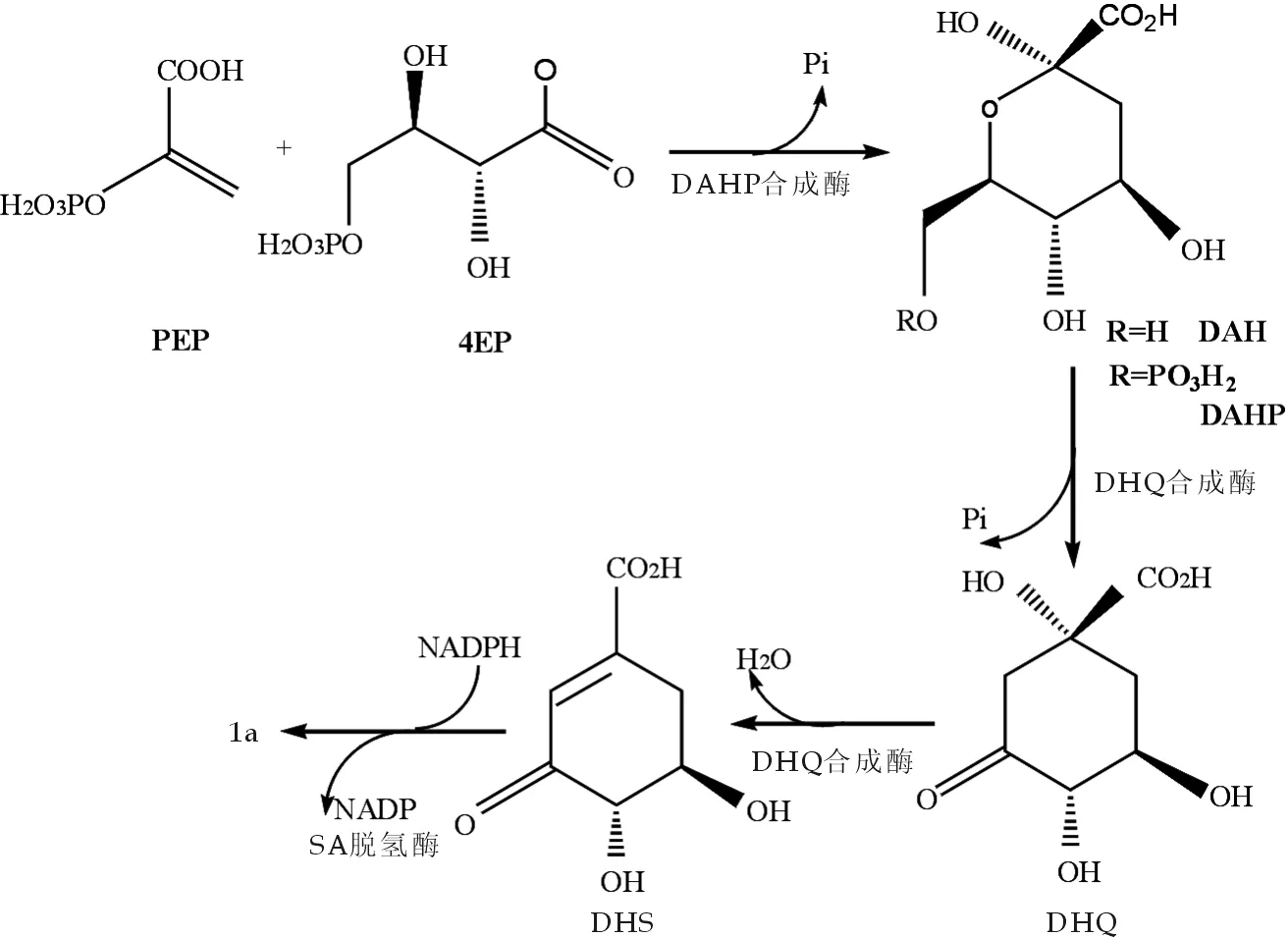
图11 莽草酸的生物合成途径
参考文献
[1] Richard Payne.Isolation of shikimic acid from Star Aniseed[J].Journal of Chemical Education,2005,82(4):599-600.
[2] Javier Adrio,Juan C. Carretero,Jose L.Garcia Ruano,etal.Enantioselective synthesis of (+)-shikimic acid and (+)-5-epi-shilkimic acid by asymmetric Diels-Alder reaction of (S)-a-sulfinylacrylates[J].Tetrahedron.,1997,8(10):1 623-1 631.
[3] Smissman, Edward E., Suh, John T.,etal. A stereospecific synthesis of dl- shikimic acid[J].J Am Chem Soc,1959,(81):2 909-2 910.
[4] McCrindle, R., Overton, K. H., Raphael, R. A.The configuration of intermediates in the total synthesis of D-(-)- shikimic acid[J]. Tetrahedron Lett, 1968,15:1 847-1 849.
[5] Bestmann, Hans J., Heid, Hans A.Stereospecific synthesis of optically pure quinic acid and shikimic acid from D-arabinose[J].Angew Chem Int Ed Eng,1971,10(5):336-337.
[6] Kamikubo, Takashi, Ogasawara,etal.A new stereocontrolled route to (-)- shikimic acid[J]. Chemistry Letters,1996, (11):987-988.
[7] A concise enantio- and diastereo-controlled synthesis of (-)-quinic acid and (-)-shikimic acid[J]. Chemical Communications (Cambridge),1998, (18):2 033-2 034.
[8] Naoyuki Yshida,Kunio Ogasawara. An enantioconvergent route to (-)-shikimic acid via a palladium-mediateed elimination reaction[J].Orangnic Letter,2000,2(10):1 461-1 263.
[9] Javier Adrio,Juan C.Carretero. Enantioselective synthesis of (+)-shikimic acid and (+)-epi-shikimic acid by asymmetric Diels-Alder reaction of (S)-sulfinylacrylates[J].Tetrahedron,1997,(8):1 623-1 631.
[10] Surachai Pornpakakul,Robin G. Asymmetric synthesis of (-)-4-epi-shikimic acid[J]. Tetrahedron Lett,2000,(41): 2 691-2 694.
[11] George W.J.Fleet,Tony K. M, Shing.An Entry to chiral cyclohexenes from carbohydrates:A short ,efficient, and enantiospecific synthesis of (-)-shikimic acid from D-Mannose[J].J.Chem.Soc,Chem.Commum,1983,(3):849-850.
[12] Shende jiang,Kevin. Enantiospecific synthesis of (-)-5-epi-shikimic acid and (-)-shikimic acid[J]. J. Chem. Soc, Perkin Trans 1,Organic and Bio-Organic Chemistry, 1997, (12), 1 805-1 814.
[13] Preparation of (-)-shikimic acid and its precursors[P]. Jpn. Kokai Tokkyo Koho ,1999.
[14] Pawlak, John L., Berchtold, Glenn A. Total synthesis of (-)-chorismic acid and (-)- shikimic acid[J]. J Org Chem, 1987, 52 (9): 1 765-1 771.
[15] Ryoyasu Saijo,Tadakazu Takeo.Some properties of the initial four enzymes involved in shikimic acid biosynthesis in tea plant[J].Agric.Biol.Chem,1979,43(7):1 427-1 432.
[16] Sunil S.Chandran,Jian Yi, K.M.Draths,Ralph von Daeniken,etal.Phosphoenolpyruvate availability and the biosynthesis of shikimic acid[J].Biotechnol.Prog, 2003,19 (3) :808-814.
[17] Louise Johansson,Anna Lindskog,Gustav Silfversparre,etal.Shikimic acid production by a modified strain ofE.coli(W3110.shil 1) under phosphate-limited and carbon-limited conditions[J].Biotechnology and Bioengineering, 2005,92 (5) :541-552.
[18] Marco Kramer, Johannes Bongaerts, Roel Bovenberg,etal.Metabolic engineering for microbial production of shikimic acid[J].2003,5:277-283.
