O-GlcNAcase抗原片段的选择、优化表达和多克隆抗体的制备
2011-02-09林霖李国超李中华田高飞李静刘艳玲
林霖,李国超,李中华,徐䶮,田高飞,李静,刘艳玲
1 南开大学药学院 元素有机化学国家重点实验室,天津 300071
2 莱芜职业技术学院信息工程系,莱芜 271100
N-乙酰葡萄糖胺 (O-GlcNAc) 糖基化是近年来发现的一种通过O-糖苷键将单个N-乙酰氨基葡萄糖(GlcNAc) 连接到蛋白质丝氨酸/苏氨酸 (Ser/Thr) 羟基上的蛋白质翻译后修饰[1]。该修饰主要发生于细胞核和细胞质中,是一种动态、可诱导、可调控的修饰方式,这一特性与蛋白质的磷酸化相似[2]。它广泛存在于真核细胞及能侵染真核生物的病毒中,现已发现1 000多种O-GlcNAc修饰的蛋白,如微管相关蛋白 Tau、抑瘤因子 p53、胰岛素受体底物蛋白IRS-1等,从而在基因转录、蛋白翻译、信号传导、细胞周期调控和应激反应中起着重要的作用[2-3],因此O-GlcNAc代谢的失衡将导致神经退行性疾病、II型糖尿病、癌症等许多重大疾病的发生[4]。
O-GlcNAcase (OGA) (EC 3.2.1.52) 是一种存在于细胞质、细胞核中的己糖胺酶,在哺乳动物的各种组织中均有存在,但是在脑、胎盘、胰腺中的丰度最高[5]。1994年,Dong等在大鼠脾脏中提取纯化出该种酶,并被命名为氨基己糖苷酶 C (Hexosaminidase C,HEXC)[6];2001年Gao等在牛脑和人脑中克隆出其基因序列[5]。一级序列分析显示,OGA基因与 1998年发现的脑膜瘤表达抗原MGEA5基因序列完全相同,定位于人10号染色体(10q24.1~q24.3),而这一区域与Alzheimer疾病密切相关,另外也有证据表明 MGEA5基因的单核苷酸多态性与墨西哥人群Ⅱ型糖尿病的发生相关[5]。将线虫OGA编码基因敲除以后,线虫的核孔蛋白和大量胞质蛋白的 O-GlcNAc水平增加,糖原合成酶激酶3-β (Glycogen synthase kinase 3-β,GSK 3-β) 的表达增加,糖原和海藻糖的积累增加,脂类储存下降,体内代谢和信号传递呈现非胰岛素依赖的Ⅱ型糖尿病模式[7]。OGA的基因定位及基因敲除线虫的表型表明,OGA与Alzheimer疾病及Ⅱ型糖尿病密切相关,该酶的调节可能涉及二者的发病机理[8]。
本课题组Li等对人源OGA存在的变体进行了研究,包括全长OGA (fOGA,full-length OGA)、变体 OGA (vOGA,spliced variant OGA) 和小片段OGA (sOGA,shortest OGA),它们都存在一共有的保守糖苷酶区域,另外,fOGA的C端与乙酰基转移酶有较高的同源性 (图1)[9]。然而针对各变体的定位、调控及对目标蛋白识别机制的研究报道较少。

图1 人OGA的三种变体[9]Fig. 1 Three isoforms of human OGA[9].
fOGA由916个氨基酸组成,在大肠杆菌中表达后,大部分以包涵体的形式存在,使用切胶回收后的蛋白作为抗原,步骤繁琐,且成功率较低[10]。而使用多肽偶联载体蛋白的方法制备的抗体特异性较差[11]。因此,至今未有商业化OGA抗体的出现,限制了OGA代谢调控及相关生物学研究。为了更好研究OGA的生物学作用,本实验对OGA进行抗原性和亲水性分析,选择N端1~350 aa (sOGA) 作为抗原,在大肠杆菌中进行表达;经过多种条件优化,我们获得了大量可溶性好、纯度高的 sOGA;并以此免疫新西兰大耳白兔制备OGA的多克隆抗体。抗体的ELISA和Western blotting检测结果表明该抗体可以有效、特异识别OGA的多种变体,可以应用于OGA的生物学研究中。
1 材料与方法
1.1 材料
质粒 pET-28a 和大肠杆菌 DH5α、BL21(DE3)均为本实验室保存。人源 OGA cDNA (GenBank Accession No. AB014579) 重组质粒 pET32a-fOGA由美国 Johns Hopkins大学 G.W.Hart教授惠赠。DNA 限制性内切酶NotⅠ和XhoⅠ、T4 DNA连接酶购自大连 TaKaRa生物工程有限公司。Ex Taq DNA聚合酶、Agarose Gel DNA Purification Kit、MiniBEST Plasmid Purification Kit购自TIANGEN公司。1 kb DNA Ladder Marker购自Fermentas公司。宽范围蛋白 Marker购自 NEB公司。酵母提取物(Yeast extract) 和胰化蛋白胨 (Tryptone) 购自Oxoid公司。HRP标记的羊抗兔 IgG 抗体购自CWBIO公司,弗氏完全及不完全佐剂为Pierce公司产品。CnBr活化的Sepharose 4B Ni-NTA亲和柱及Superdex75分子筛柱为 GE Healthcare公司产品。PVDF薄膜为Osmonics公司产品,PCR引物及重组质粒测序由华大基因完成。其余试剂均为国产分析纯。试验动物新西兰大耳白兔2只,体重约2.0 kg,购于北京市海淀区兴隆实验动物养殖厂。
1.2 引物的设计和sOGA片段的扩增
根据人OGA基因全长cDNA序列,设计N端1~350 aa (sOGA) 原核表达引物,序列如表1所示。以质粒pET32a-fOGA为模板进行PCR扩增,产物用1.0%琼脂糖凝胶电泳检测,切胶回收目的片段。
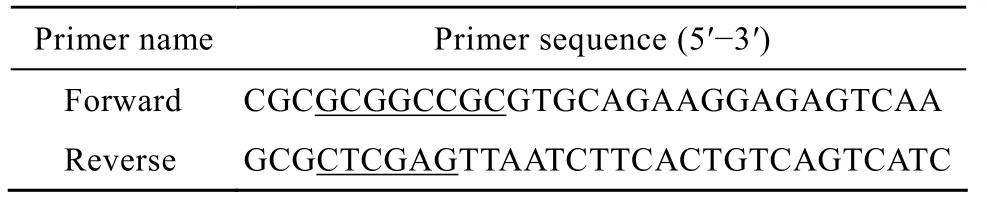
表1 引物序列Table 1 Primer sequence
1.3 重组表达载体的构建
用限制性内切酶 NotⅠ和 XhoⅠ酶切验证回收后的 PCR产物和质粒 pET-28a,将 sOGA片段和pET-28a酶切产物用1.0%琼脂糖凝胶电泳纯化回收后,分别取4 µL sOGA基因与2 µL载体pET-28a,用T4 DNA连接酶于16 ℃连接过夜,连接后的产物转化至大肠杆菌 DH5α感受态细胞,接种于含50 mg/mL卡那霉素的LB平板上培养,次日挑取单克隆菌扩大培养后,试剂盒提取质粒 DNA,利用双酶切和PCR鉴定重组质粒pET-28a-sOGA,酶切和PCR扩增后的产物用1.0%琼脂糖凝胶电泳进行分析[12]。
1.4 抗原片段的优化表达和纯化
将阳性克隆接种于5 mL含有50 mg/mL卡那霉素的LB培养基中,37 ℃、200 r/min过夜培养。次日,吸取100 µL培养物加入到10 mL的新鲜培养基中。37 ℃、200 r/min培养至OD600=0.6~0.8左右,添加IPTG至终浓度为0.01~0.05 mmol/L,13 ℃、110 r/min继续培养5~15 h,离心收集菌体,超声波破碎后采用SDS-PAGE电泳检测蛋白表达情况[12]。
利用优化好的IPTG浓度和诱导时间,进行大量培养。诱导后的菌液在4 ℃、8 000 r/min 离心20 min,收集菌体。按照每克湿菌体加入10 mL PBS (pH 7.4) 柱平衡缓冲液的量充分悬浮菌体,在冰浴条件下超声破碎菌体 (160 W,工作4 s,间歇7 s,共99次),然后4 ℃、16 000 r/min 离心30 min,收集上清液。将上清液加至预先用平衡缓冲液充分平衡好的Ni-NTA亲和层析柱 (1 mL),并用平衡缓冲液淋洗10个柱体积,然后换用含不同浓度咪唑的平衡缓冲液洗脱sOGA蛋白,收集各组分。采用超滤法 (Millipore,截留分子量10 000 kDa) 对纯化后的蛋白更换缓冲液至低盐PBS (pH 7.4) 进行浓缩后,使用 FPLC purifier系统将蛋白样品上样至superdex75 (120 mL) 柱进行分子筛层析纯化。各组分取样进行12% SDS-PAGE电泳分析。蛋白浓度采用BCA法测定。
1.5 OGA糖苷酶酶活测定
酶活测定使用Macauley等[13]的方法。在缓冲液(50 mmol/L NaH2PO4;100 mmol/L NaCl和0.1%牛血清白蛋白,pH 6.5) 中,以4-MU-GlcNAc作为荧光底物进行酶活检测。添加1~3 µL酶的反应混合物,在37 ℃下孵育4~30 min,然后加入150 µL的200 mmol/L甘氨酸 (pH 10.75) 来终止酶促反应。使用CARY Eclipse型荧光分光光度计96孔板系统,在激发波368 nm,发射波450 nm条件下进行检测,测定不同稀释浓度 4-MU (4-methylumbelliferone)的荧光值,制作标准曲线,将检测获得的酶促反应的荧光值以此为对照进行计算,所有的实验都在相同条件下重复3次。
1.6 OGA多克隆抗体血清的制备及效价测定
首次免疫将纯化后的sOGA溶液加等体积弗氏完全佐剂乳化后,皮下多点注射。第2 (第21天)、3次 (第35天) 免疫目的蛋白与等体积弗氏不完全佐剂混合,皮下多点注射。免疫10 d后,每只兔子耳缘静脉取血0.5~1 mL,分离抗血清,间接ELISA检测免疫后血清效价。
用纯化的重组sOGA包被酶标板 (0.2 µg/孔),4 ℃冰箱过夜;PBST洗涤3次 (每次振板5 s);5%脱脂奶封闭 (300 µL/孔,37 ℃,封闭 2 h);PBST洗涤 3次;将待检测抗血清按设计稀释度(1∶1 000、1∶3 000、1∶9 000、1 ∶2 7 000、1∶ 81 000、1 ∶ 243 000,1 ∶ 729 000) 稀释后加入酶标板中,100 µL/孔,37 ℃孵育1 h;阴性对照为免疫前血清,按1∶1 000倍稀释,空白对照为PBS、PBST洗涤;将HRP标记的山羊抗兔 IgG (1∶10 000) 稀释后加入酶标板,100 µL/孔,37 ℃,孵育40 min,PBST洗涤,TMB显色,酶标仪测450 nm波长的OD值。
效价达到指标后,以心脏穿刺的方法采全血,分离抗血清。
1.7 OGA多克隆抗体的纯化及效价测定
抗原亲和柱制备:用活化缓冲液将 CnBr Sepharose 4B充分活化,平衡活化的CnBr凝胶至基线平稳,将抗原sOGA加入平衡好的凝胶中,常温旋转反应2 h,加入封闭缓冲液常温旋转反应1 h,封闭剩余活化基团,用洗脱缓冲液洗柱。
特异性抗体纯化:用结合缓冲液平衡抗原亲和柱至基线平稳,将抗血清负载上柱,收集流穿液;将流穿液再次上柱,继续平衡至基线平稳,加入洗脱缓冲液洗脱,收集洗脱峰,SDS-PAGE检测纯度。用10 mmol/L PBS (pH 7.2) 透析收集的洗脱液,浓缩透析后的抗体,测定浓度。
效价测定:用纯化的sOGA蛋白包被酶标板,用间接ELISA技术 (同上) 测定抗体效价。
1.8 OGA多克隆抗体的Western blotting分析
蛋白样品经SDS-PAGE分离后转移至PVDF膜。5%脱脂牛奶 (溶于TBST中) 4 ℃ 封闭过夜,TBST洗膜后加入抗OGA多克隆抗体 (1∶5 000),室温反应1.5 h。TBST (25 mmol/L Tris-HCl,pH 7.5;50 mmol/L NaCl,0.1% Tween 20) 洗涤3次,10 min/次,1∶3 000加入HRP标记的羊抗兔IgG抗体,室温孵育1.5 h,TBST洗涤3 次,10 min/次,洗涤后加 ECL使 X光胶片感光,检测抗体的特异性[12]。
2 结果与分析
2.1 人源OGA基因片段 (aa1-350,sOGA) 的获得和载体构建
以人源 OGA cDNA为模板,设计针对 N端 1~350 aa的引物进行PCR扩增,经过PCR扩增后的产物进行1.0%琼脂糖凝胶电泳分析,PCR扩增后的产物大小约为1 050 bp (图2),与sOGA基因的理论大小相符合。
将回收后的PCR产物采用限制性内切酶NotⅠ和 XhoⅠ酶切,然后与经过 NotⅠ和 XhoⅠ酶切后的表达载体 pET-28a连接,转化至大肠杆菌 DH5α感受态细胞中,挑取单菌落提取质粒,采用NotⅠ和XhoⅠ进行双酶切鉴定 (图2),酶切后的质粒在相同位置也出现大小一致的片段,说明目的基因片段与表达载体连接正确,表达载体pET-28a-sOGA的构建是成功的。DNA序列测定结果证实:扩增序列与人源OGA cDNA序列一致。
2.2 sOGA的优化表达和纯化
通过控制不同IPTG浓度、不同诱导时间等因素对sOGA (1~350 aa) 片段进行诱导表达条件的优化,使其在尽可能少形成包涵体的情况下有较高的可溶性表达。通过SDS-PAGE电泳 (图3A) 分析,发现诱导后的sOGA在45 kDa处出现一条带,与预期大小一致。同时确定sOGA的最佳诱导表达条件分别为:0.05 mmol/L IPTG,13 ℃,110 r/min,10 h。
按照优化后的诱导表达条件,进行扩大培养。收集菌体后经过超声破碎、高速离心,将上清液过 Ni亲和层析柱和分子筛层析进行纯化,经 SDS-PAGE电泳和考马斯亮蓝染色检测,结果表明实验获得了纯度较高的His标签融合sOGA蛋白 (纯度>95%),产率达8 g/L (图3B)。
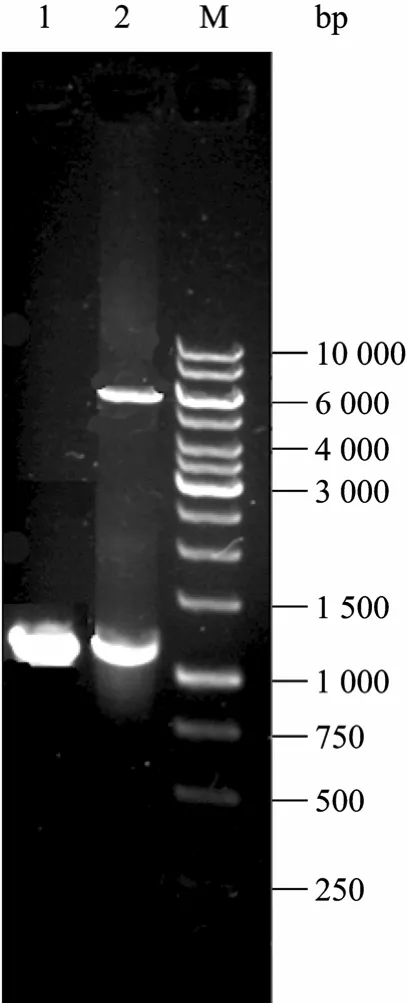
图2 表达载体的构建及酶切鉴定Fig. 2 Construction and digestion analysis of pET-28a-sOGA recombinant plasmid. 1: PCR result of sOGA; 2: pET-28a-sOGA digested with Not I and Xho I; 3: DNA marker (GeneRuler TM 100 bp).
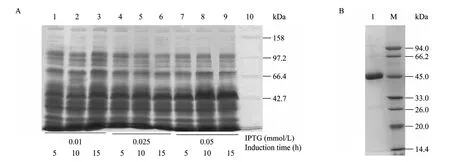
图3 sOGA的优化表达和纯化Fig. 3 Optimized expression and purification of sOGA. (A) 1: 0.01 mol/L IPTG, 5 h; 2: 0.01 mmol/L IPTG, 10 h; 3: 0.01 mmol/L IPTG, 15 h; 4: 0.025 mmol/L IPTG, 5 h; 5: 0.025 mmol/L IPTG, 10 h; 6: 0.025 mmol/L IPTG, 15 h; 7: 0.05 mmol/L IPTG, 5 h; 8: 0.05 mmol/L IPTG, 10 h; 9: 0.05 mmol/L IPTG, 15 h; 10: protein marker. (B) 1: sOGA; 2: protein marker.
2.3 sOGA糖苷酶学特性检测
由于 Ni2+纯化洗脱液含有较多的咪唑,可能影响后期酶活测定结果。因此我们选用超滤管更换缓冲液,将酶更换至新缓冲液 (50 mmol/L Tris-HCl,pH 7.0) 中,然后利用系列浓度(5 mmol/L,2.5 mmol/L,1.25 mmol/L,0.625 mmol/L,0.312 5 mmol/L,0.156 25 mmol/L) 的底物4-MU-OGlcNAc,测定sOGA片段的Lineweaver-Burk曲线(如图4),经测定sOGA的Km为1.84 mmol/L,催化活性为:106 nmol/(min·mg)。表明sOGA片段是糖苷酶活性中心,可以以此片段为抗原制备 OGA抗体。
2.4 OGA多克隆抗体的制备、纯化和效价检测
利用抗原活化了的Sepharose 4B微珠与抗体结合的特性,从免疫后的兔血清中纯化IgG,取10 µL纯化后的OGA多克隆抗体,进行SDS-PAGE电泳检测 (图5),考马斯亮蓝染色后发现有一条50 kDa的条带,定量测定纯化后的抗体浓度为3.75 mg/mL。
用间接ELISA方法对亲和纯化后的抗体进行了效价检测 (表2),鉴于OD450值为阴性对照孔OD450值的2.1倍以上即为阳性的原则,认为sOGA抗原亲和纯化兔多克隆抗体的检测灵敏度0.11 ng/mL,抗血清的稀释度为1∶80 000。
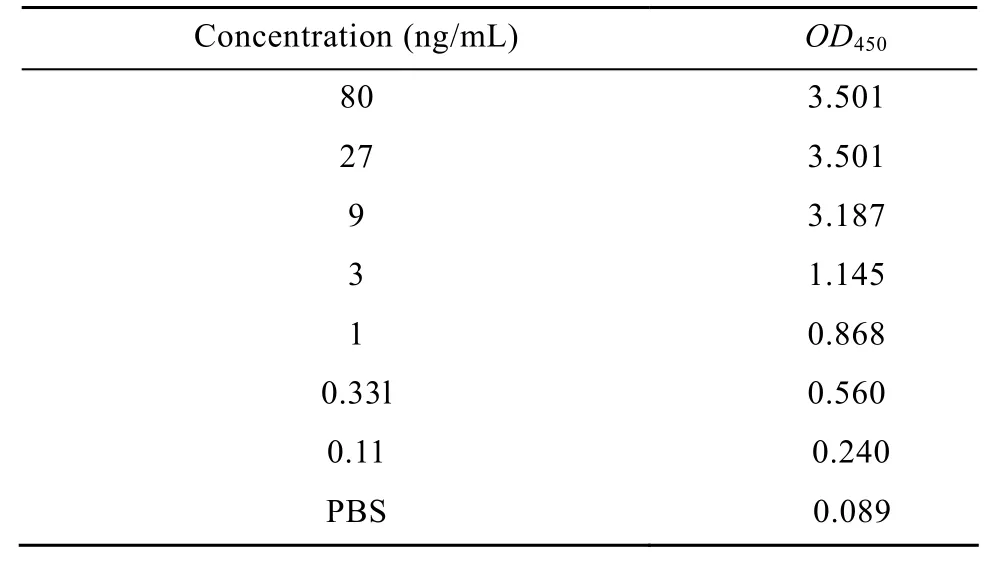
表2 ELISA检测抗体效价Table 2 ELISA analysis of the titer of OGA polyclonal antibody
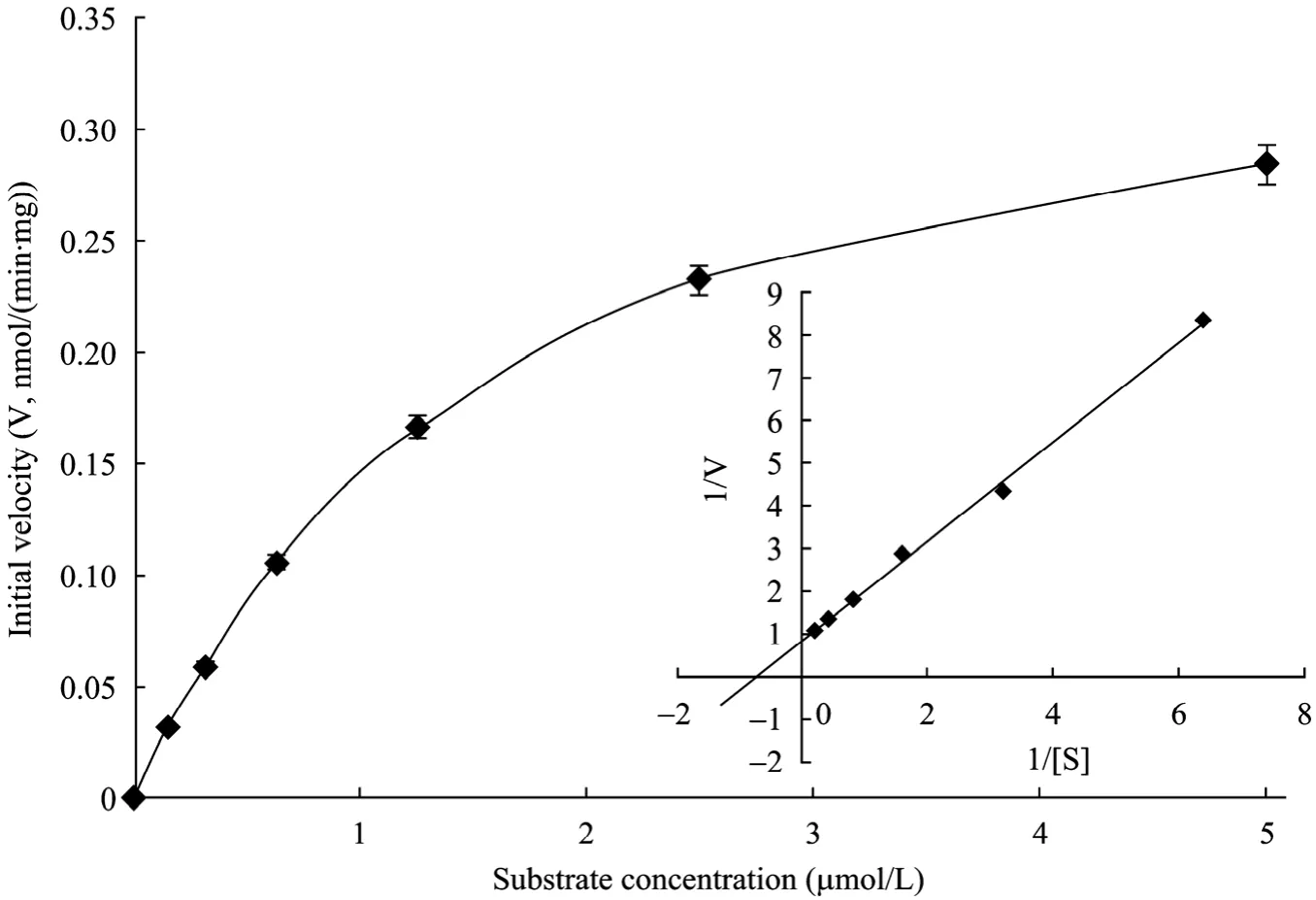
图4 sOGA的酶活测定和Lineweaver-Burk测定Fig. 4 An activity curve and a Lineweaver-Burk plot were generated by varying concentrations of substrate (4-MU-GlcNAc).
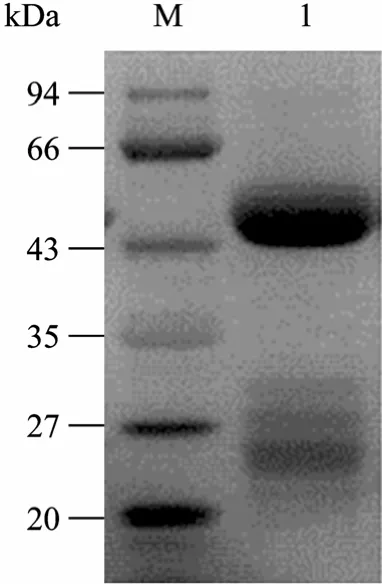
图5 亲和纯化后的OGA多克隆抗体Fig. 5 Purified Anti-sOGA polyclone antibody. M: protein marker; 1: anti-sOGA polyclone antibody.
2.5 OGA多克隆抗体特异性检测
为检测纯化后OGA多克隆抗体的特异性,分别取过表达不同人源OGA变体蛋白fOGA (1~916 aa),vOGA (1~677 aa),sOGA (1~350 aa) 的大肠杆菌细胞裂解液50 µg 上样,进行SDS-PAGE (10%) 分离,用制得的抗体进行免疫印迹检测 (图 6)。结果发现,不同 OGA变体在相应大小 (150 kDa,98 kDa,45 kDa) 处均检测到特异性条带,而转化空质粒的大肠杆菌细胞裂解液未有明显的杂交带,表明抗体具有很好的特异性,并能识别不同变体的OGA,可以应用于OGA的细胞生物学研究。
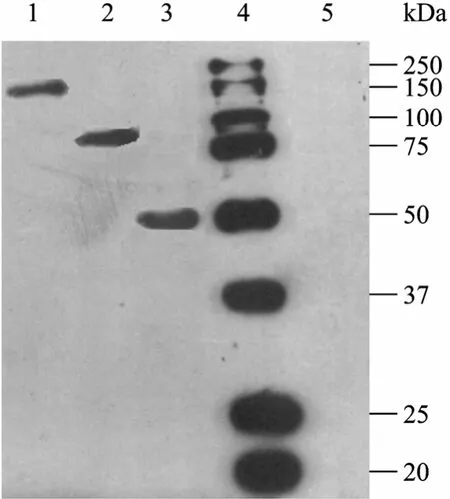
图6 抗OGA多克隆抗体的特异性检测Fig. 6 Identification of the specificity of OGA polyclonal antibody. 1: fOGA; 2: vOGA; 3: sOGA; 4: protein marker; 5: control.
3 讨论
人源O-GlcNAcase在细胞中存在2种不同大小的变体,全长 (fOGA) 含有916个氨基酸,存在2个结构域:氨基端约 (1~350 aa) 具有β-N-乙酰葡萄糖苷酶活性,羧基端 (活性中心约为592~916 aa) 被推测可能具有组蛋白乙酰基转移酶 (HAT) 活性;而变体 (vOGA) 由于剪切机制不同,只含有N端677个氨基酸,缺少HAT结构域。sOGA是含有350个氨基酸的最小糖苷酶活性中心。由于缺少有效的OGA抗体,3种变体OGA的调控及相关疾病机理的研究受到较大的限制。
我们先前的研究表明,fOGA的N端1~350 aa (sOGA),在大肠杆菌中易于表达纯化,并且软件预测发现该片段具有较好的免疫原性和亲水性。因此,我们选取sOGA作为抗原。首先将其构建到N端含有6个His标签的原核表达载体pET-28a中;设立不同的诱导条件优化pET-28a-sOGA的表达;然后选择可溶性表达最好的条件进行大量表达;Ni柱亲和纯化和分子筛层析后,获得了大量高纯度的sOGA蛋白;通过测定sOGA的糖苷酶活,确定该片段是糖苷酶活性中心,可以用来制备OGA变体的抗体。最后以其作为抗原免疫兔子制备得到了 OGA多克隆抗体。抗体经亲和纯化后,ELISA法测定抗体效价较高 (1∶80 000);Western blotting方法检测到,不同大肠杆菌表达的重组OGA变体 (fOGA、vOGA和 sOGA),均可以与制备抗体产生预期杂交特异带。实验结果表明,以sOGA为抗原制备的抗体,可以高效、特异地识别OGA变体。
OGA是O-GlcNAc糖基化修饰中的关键酶,业已证明该酶的调控可能与 Alzheimer疾病及Ⅱ型糖尿病的发病机理相关[15-17]。Tau蛋白的异常过度磷酸化是Alzheimer疾病过程中的关键事件[3],然而最近研究发现 Tau蛋白除被磷酸化修饰外,也被O-GlcNAc糖基化修饰 (大约 12个位点:Thr231、Ser262、Ser396、Ser422等),且这些位点的O-GlcNAc修饰与磷酸化存在竞争性的关系[3],因此OGA活性的降低或失调可能是导致该疾病的原因。
本实验制备的 OGA多克隆抗体可以应用于Alzheimer疾病的生物学试验中,为深入研究 OGA的调控机制及功能奠定了基础。
REFERENCES
[1] Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes: evidence for O-linked GlcNAc. J Biol Chem, 1984, 259(5): 3308−3317.
[2] Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science, 2001, 291(5512): 2376−2378.
[3] Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature, 2007, 446(7139): 1017−1022.
[4] Zeidan Q, Hart GW. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci, 2010, 123: 13−22.
[5] Gao Y, Wells L, Comer FI, et al. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytasolic beta-N-acetylglucosaminidase from human brain. J Biol Chem, 2001, 276: 9838−9845.
[6] Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-Glucosaminidase from rat spleen cytosol. J Biol Chem, 1994, 269(30): 19321−19330.
[7] Forsythe ME, Love DC, Lazarus BD, et al. Caenorhabditis elegans ortholog of a diabetes susceptibility locus: oga-1 (O-GlcNAcase) knockout impacts O-GlcNAc cycling, metabolism, and dauer. Proc Natl Acad Sci USA, 2006, 103(32): 11952−11957.
[8] Mondoux MA, Krause MW, Hanover JA. C elegans genetic networks predict roles for O-GlcNAc cycling in key signaling pathways. Curr Signal Transd Ther, 2010, 5(1): 60−73.
[9] Li J, Huang CL, Zhang LW, et al. Isoforms of human O-GlcNAcase show distinct catalytic efficiencies. Biochemistry (Moscow), 2010, 75(7): 938−943.
[10] Crawford GL, Hart GW, Whiteheart SW. Murine platelets are not regulated by O-linked β-N-acetylglucosamine. Archives Biochem Biophys, 2008, 474(1): 220−224.
[11] Lee TN, Alborn WE, Knierman MD, et al. The diabetogenic antibiotic streptozotocin modifies the tryptic digest pattern for peptides of the enzyme O-GlcNAcselective N-acetyl-β-d-gluco saminidase that contain amino acid residues essential for enzymatic activity. Biochem Pharmacol, 2006, 72(6): 710−718.
[12] Sambrook J, Frisch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. New York: Cold Spring Harbor Laboratory Press, 1989: 19−20.
[13] Macauley MS, Whitworth GE, Debowski AW, et al. O-GlcNAcase uses substrate-assisted catalysis Kinetic analysis and development of highly selective mechanisminspired inhibitors. J Biol Chem, 2005, 280: 25313−25322.
[14] Butkinaree C, Cheung WD, Park S, et al. Characterization of β-N-Acetylgluco saminidase cleavage by caspase-3 during apoptosis. J Biol Chem, 2008, 283(35): 23557−23566.
[15] Brimble, S, Wollaston H, Edith ET, et al. The role of the O-GlcNAc modification in regulating eukaryotic gene expression. Curr Signal Transduct, 2010, 5(1): 12−24.
[16] Dentin R, Hedrick S, Xie JX, et al. Hepatic glucose sensing via the CREB coactivator CRTC2. Science, 2008, 319(5868): 1402−1405.
[17] Kang JG, Park SY, Ji S, et al. O-GlcNAc protein modification in cancer cells increases in response to glucose deprivation through glycogen degradation. J Biol Chem, 2009, 284: 34777−34784.
