鸟嘌呤四链体中Na+的移动
2010-11-30杨忠志
郭 慈 刘 翠 杨忠志
(辽宁师范大学化学化工学院,辽宁大连 116029)
EvdW和Eelec是非键作用势能.EvdW描述非键原子间的vdW相互作用,表达式为:
鸟嘌呤四链体中Na+的移动
郭 慈 刘 翠 杨忠志*
(辽宁师范大学化学化工学院,辽宁大连 116029)
Na+-G-四链体复合物是一个明显的极化体系,其形成或解离过程中,Na+的移动路线目前还不十分明确. σπ水平的原子-键电负性均衡方法融合进分子力学(ABEEMσπ/MM)模型除原子位点外,还明确地定义了孤对电子、σ键和π键的位置,并且各位点电荷随分子环境改变而浮动,因此能更好地反映该体系的极化现象.本文应用ABEEMσπ/MM方法研究了Na+-G-四平面复合物的性质,包括它的几何构型、电荷分布和结合能等,并在MP2/6-31G(d,p)水平上做了相应的从头算,两种结果十分吻合.Na+的存在改变了G-tetrad的氢键方式.通过比较Na+各条移动路线中体系的结合能,预测G-四链体中三个Na+最有可能沿α方向依次移出.以上研究为进一步应用ABEEMσπ/MM模型进行G-四链体中离子交换通道的动力学模拟打下坚实的基础.
G-四链体;Na+-G-四平面;ABEEMσπ/MM方法;从头算;移动路线
端粒是染色体末端一种特殊的、富含鸟嘌呤的结构.多项研究[1-7]发现,在某些离子条件下,富含鸟嘌呤端粒的尾链,可以通过分子内折叠或分子间相互作用,形成特殊的DNA二级结构,即鸟嘌呤四链体(G-quadruplex,G4).此结构由三个平行排列的G-四平面堆叠而成;每个G-四平面(G-tetrad)由4个鸟嘌呤通过分子间氢键连接构成,离子存在于G4空穴中.G4的形成与端粒酶引起的端粒延长有关,可抑制端粒酶发挥作用,干扰端粒复制,破坏端粒功能,对细胞生长、分化、凋亡等基本生命活动产生影响[8-9],G4的特殊结构与性质使之成为药物设计的理想方向[10-15].
已有文献[16-19]对G4进行了研究,包括结构、稳定性和G4中离子交换等相关问题.研究表明:与金属离子相互作用对G4的形成至关重要,离子的作用就是稳定G4.G4的稳定性和结构随它含有的离子类型而定,也就是说,G4对离子具有选择性.Gu等[20]研究了G4对离子的选择,表明在气相中,单价阳离子稳定G4的顺序是Li+>Na+>K+,而在水溶液中顺序恰好相反.Ma等[21]研究了亲脂性的G4中离子交换的问题,指出:并不是所有离子的结合位置都是均等的.然而,G4形成或解离过程中离子的移动路线目前还不是十分明确.要判断G4中离子的移动路线,必须要研究离子与G4的相互作用.对此本文应用从头算(MP2)[22]和ABEEMσπ/MM[23-30]方法,以一个Na+和1片G-tetrad(Na+-G-tetrad)的相互作用为切入点,验证了ABEEMσπ/MM方法的准确性.在此基础上,应用ABEEMσπ/MM方法研究了两片G-tetrad和Na+(Na+-G-tetrads)的相互作用,通过比较Na+移动过程中体系的结合能,预测了G4中Na+的移动路线.
1 计算方法
1.1 从头计算方法
在量子化学计算方法中,通常用来计算能量的有HF、DFT、MP2及CCSD[31]等方法.其中HF方法没有考虑电子相关效应,计算出的能量不够准确; DFT方法虽考虑了电子相关效应,但不能很好地处理弱键相互作用;CCSD方法虽然计算能量比较准确,但由于计算机条件限制,只能做十几个原子的体系.G4是一个相对较大的体系,考虑到MP2方法的计算精度,以及该方法节省计算机时,本文采用MP2方法计算能量,基组为6-31G(d,p).由于本文的初始构型是实验构型,因此没有进行几何优化,在计算能量的过程中应用 counterpoise方法进行了BSSE校正.
1.2 ABEEMσπ/MM模型
ABEEMσπ力场中,分子的总能量EABEEMσπ表示为:
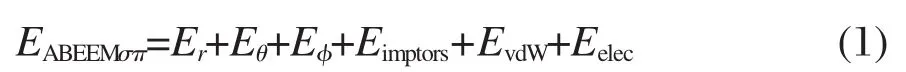
其中Er是键伸缩振动势能;Eθ是分子中连续键连的三个原子形成的键角弯曲振动势能;Eφ和Eimptors是分子中连续键连的四个原子形成的二面角扭转势能和非共面扭转势能.它们的表达式如式(2-5)所示:

其中kr和kθ表示键伸缩和键角弯曲势能的力常数, r、θ和φ是实际的键长、键角和二面角值,req和θeq表示平衡键长和键角值,V1、V2、V3和ν为二面角扭转势能项及非共面扭转势能项的展开力常数.
EvdW和Eelec是非键作用势能.EvdW描述非键原子间的vdW相互作用,表达式为:
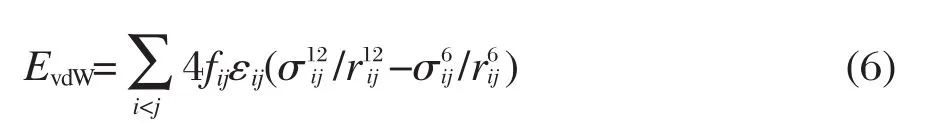
f为常数,ε为势阱深,σ为碰撞直径.此处采用了标准的联合规则:εij=(εiiεjj)1/2,σij=(σiiσjj)1/2.对于分子内相互作用,任何1-2和1-3关系的i-j原子对,fij=0.0,任何1-4关系的i-j原子对,fij=0.5,所有其它情况下fij=1.0.
静电相互作用项 Eelec中的电荷计算是ABEEMσπ模型的精华所在,公式如下:

其中,qi和qj是位点i和j的电荷,rij是位点i和j的距离,当i和j之间的最短连接路径关系(包括σ键位点)小于1-6时,kij=0;当i和j在氢键相互作用区域时,kij=kH-bond(氢键拟合函数);其它所有情况,kij= 0.57.在ABEEMσπ模型中,σ键处于两成键原子共价半径之比处;π键处于垂直于双键所在平面,置于双键原子上下两侧共价半径处;孤对电子处于距离双键原子共价半径处.
1.3 ABEEMσπ/MM模型参数的确定
ABEEMσπ参数的拟合是计算所有性质的一个关键步骤.本文通过线性回归和最小二乘法优化确定参数.参数的拟合不仅产生与从头计算相一致的电荷分布、结合能,而且获得了与实验构型相一致的几何构型[32].其中G4体系的范德华参数列于表1.

表1 ABEEMσπ/MM模型中G4体系的vdW参数Table 1 vdW parameters of G4 in ABEEMσπ/ MM model
如果想要解释广泛的化学现象,那么基于静电力场的分子模拟中,包含氢键效应是至关重要的. G4体系中鸟嘌呤间有两种类型的氢键:(1)O6(原子编号见图1中所示)原子上的孤对电子与其相邻鸟嘌呤上H45原子之间形成的氢键,或O6原子上的孤对电子与其相邻鸟嘌呤上H47原子之间形成的氢键;(2)N3原子上的孤对电子与其相邻鸟嘌呤上H47原子之间形成的氢键.这两种类型的氢键拟合函数是沿用ABEEMσπ/MM模型原有的函数[29].本文特别关注的是Na+与鸟嘌呤中O6原子、N10原子、N3原子上孤对电子之间的静电相互作用.为了更好地描述这三种静电相互作用,我们用ABEEMσπ/MM模型拟合了在MP2/6-31G(d,p)水平上的两个静电区域处于不同距离时的结合能,使二者结果有很好的一致性.从而引入三个可调参数k(RlpO=,Na+),k(RlpN10,Na+)和k(RlpN3,Na+).这些参数随Na+与孤对电子之间距离的变化而改变,更好地体现了该区域内的极化现象,这三个参数的表达式分别为:

其中R表示Na+与孤对电子之间的距离.
Na+与G-tetrad相互作用研究中用到的其它参数沿用Yang等人已经报道的相应参数[29-30].
2 结果与讨论
2.1 几何构型和电荷分布
应用ABEEMσπ/MM模型优化的G-tetrad与Na+-G-tetrad结构相差很大.G-tetrad构型中,四个鸟嘌呤通过交叉型氢键存在,如图1(a)所示.Na+-G-tetrad构型中,四个鸟嘌呤趋近于在一个平面内,采用平行型的氢键,Na+在四个鸟嘌呤形成空穴的下方,如图1(b)所示.分析可得,G-tetrad结构中,中心部位的四个氧原子之间存在很大的静电排斥作用,为了降低这种排斥作用,O6原子上的孤对电子与N40上的H45以及N42上的H47形成了交叉型的氢键.当有Na+存在的情况下,Na+中和了G-tetrad中心部位集中的大量负电荷,平衡了G-tetrad中心部位四个氧原子之间的静电作用,使G-tetrad采取平行型的氢键,增加了G-tetrad结构的稳定.


表2 ABEEMσπ/MM模型优化的Na+-G-tetrad的结构参数与实验数据的比较Table 2 Comparison of structural parameters for Na+-G-tetrad from experiment and optimized by ABEEMσπ/MM model
表2列出的是ABEEMσπ/MM模型优化的Na+-G-tetrad的部分结构参数与实验构型的比较. ABEEMσπ/MM模型优化得到的Na+-G-tetrad复合物的几何构型参数与实验结构参数[32]比较可知,二者的结构符合得很好.鸟嘌呤单体键长的绝对平均偏差为0.0013 nm,键角的绝对平均偏差为1.18°.从表2中可以看出,原子间距离的绝对平均偏差为0.0026 nm,角度的绝对平均偏差为2.18°,都在较合理的范围内.可见,ABEEMσπ/MM模型能够正确地预测Na+-G-tetrad的结构信息.
表3列出的是ABEEMσπ/MM模型计算的G-tetrad和Na+-G-tetrad中氧原子各位点所带的电荷. Na+-G-tetrad是一个明显的极化体系,ABEEMσπ/ MM模型除原子位点外,还明确地定义了孤对电子、σ键和π键的位置,并且随分子环境的改变,各位点的电荷发生相应的浮动,这样更好地反映了该体系的极化现象.将表中各位点的电荷回归到原子后:G-tetrad中,四个氧原子所带的电荷相等都是-0.5798e;Na+-G-tetrad中,qO6=-0.6538e;qO17= -0.6597e;qO28=-0.6623e;qO39=-0.6450e.由此可以看出,将G-tetrad和Na+-G-tetrad中各位点电荷回归到原子后,氧原子带有不同程度的负电荷.因为Na+-G-tetrad结构中Na+带有一定的正电荷,并且Na+和氧原子间存在静电相互作用,所以氧原子被极化,带更多的负电荷.通过对比可知,O的孤对电子上的电荷改变最大,而不是原子位点处.并且有Na+存在时,靠近Na+的π键位点的负电荷明显地比远离Na+的π键位点的负电荷多;没有Na+存在时O原子上下的π键位点所带电荷基本相等.
ABEEMσπ/MM模型不仅定义了非原子位点,并且引入可调参数来研究Na+与O6原子、N10原子、N3原子上的孤对电子之间的静电相互作用,所以该模型能很好地模拟G-tetrad和Na+-G-tetrad体系中的氢键和静电相互作用.总体来说, ABEEMσπ/MM模型能够合理地预测G-tetrad的结构,与其它文献报道一致[20],并且优化的Na+-G-tetrad构型与实验构型有很好的一致性,进而说明了ABEEMσπ/MM模型在描述G-tetrad和Na+-G-tetrad的几何构型方面十分可靠.

表3 ABEEMσπ/MM计算的G-tetrad和Na+-G-tetrad中氧原子各位点所带的电荷Table 3 Charges of all sites of O atoms in G-tetrad and Na+-G-tetrad computed by ABEEMσπ/MM

图2 Na+移动方向正视图(a)和俯视图(b)Fig.2 The front(a)and vertical(b)view of the moving direction of Na+
2.2 Na+和一片G-tetrad相互作用
G4形成或解离过程中,涉及到Na+能否移出G4,以及Na+在移动的过程中能否跟其它的离子交换(如K+)等问题,本文应用ABEEMσπ/MM模型初步预测了Na+的移动路线.
初始构型取自蛋白质晶体数据库,其数据库标号为1KF1[32].删去磷酸和糖环,并在碱基上加上氢原子,将其中的钾离子改为钠离子.为了检验模型的准确性,取第1片G-tetrad和Na+(2)(Na+(2)-G-tetrad)作为初始结构,如图2所示.讨论了一个Na+与一片G-tetrad的相互作用,并与MP2/6-31G(d,p)水平的从头算结果进行了对比.将Na+(2)和Na+(3)的连线方向定义为α方向;过Na+(2),且垂直于α方向取一个平面I,将第1片G-tetrad中两鸟嘌呤的夹缝方向在平面I中的投影定义为β方向;将第1片G-tetrad中两鸟嘌呤的对角线方向在平面I中的投影定义为γ方向,图中箭头所指方向为正方向.根据Na+(2)-G-tetrad结构的特点,α、β、γ三个方向是该结构中最具代表性的方向,以Na+(2)所在位置为起点(平衡位置),讨论了该离子分别沿α、β、γ三个方向的移动.结合能ΔE计算公式如下:
ΔE=E[Na+(2)]+E[G-tetrad]-E[Na+(2)-G-tetrad](11)其中E[Na+(2)]为Na+(2)的能量,E[G-tetrad]为一片G-tetrad复合物的能量,E[Na+(2)-G-tetrad]为Na+(2)-G-tetrad复合物的能量.
2.2.1 Na+(2)-G-tetrad中Na+(2)沿α方向移动
Na+(2)沿α方向的移动以X=0.1750 nm(X为Na+(2)距平面I的距离)为对称轴.Na+(2)沿α方向移动是指:第1片G-tetrad的坐标不变,改变Na+(2)距平面I的距离,如图3(a)所示.首先将Na+(2)沿α正方向,每隔0.0100 nm取一个结构;再将Na+(2)沿α负方向每隔0.0100 nm取一个结构,到0.3250 nm后每隔0.1000 nm取一个结构,到1.6250 nm后每隔0.5000 nm取一个结构,直到4.0250 nm.图3(b)展示了MP2水平和ABEEMσπ/MM模型下,Na+(2)沿α方向移动时,Na+(2)-G-tetrad体系结合能变化曲线.

图3 Na+(2)沿α方向移动的正视图(a)和结合能变化曲线(b) Fig.3 The front view(a)and the binding energy curve(b)of Na+(2)moving along the α orientationd:the distance between Na+and its equilibrium position along corresponding moving direction;The data in parentheses denote the coordinate ofextremal point for Na+(2)-G-tetrad when Na+(2)moves along the α orientation obtained from ABEEMσπ/MM model;the view of big pane is amplificatory view of small pane.
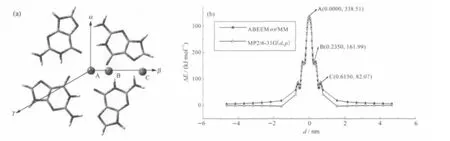
图4 Na+(2)沿β方向移动的俯视图(a)和结合能变化曲线(b)Fig.4 The vertical view(a)and the binding energy curve(b)of Na+(2)moving along the β orientationThe data in parentheses denote the coordinates of extremal points for Na+(2)-G-tetrad when Na+(2)moves along the β orientation.
根据总的结合能变化曲线可以看出,Na+(2)与平面I之间距离为0.1000 nm时(图3中A点位置),复合物Na+(2)-G-tetrad的结合能最大,结构最稳定.而实验构型中K+在平面I内时,结构最稳定.随着Na+(2)向α正方向或α负方向移动,该复合物的结合能逐渐减小,直到距离小于-1.8500 nm或大于2.0500 nm后,结合能约等于0.00 kJ·mol-1,说明Na+(2)与G-tetrad完全分开,不存在相互作用,相当于两个独立的个体.
2.2.2 Na+(2)-G-tetrad中Na+(2)沿β方向移动
由于Na+(2)沿β正负方向的移动是对称的,所以本文仅以向β正方向移动为例进行详细分析.Na+(2)沿β方向移动指:第1片G-tetrad的坐标不变,只是沿第1片G-tetrad中两鸟嘌呤的夹缝在平面I中的投影方向移动,改变Na+(2)到平衡位置的距离,如图4(a)所示.首先将Na+(2)沿β正方向每隔0.0200 nm取一个结构,到0.5150 nm后每隔0.0500 nm取一个结构,到1.5650 nm后每隔0.5000 nm取一个结构,直到4.6650 nm.图4(b)展示了MP2水平和ABEEMσπ/ MM模型下,Na+(2)沿β方向移动时,Na+(2)-G-tetrad体系的结合能变化曲线.
可以看出平衡位置时(图4中A点位置)结合能最大,此时体系最稳定.随着Na+(2)向β正方向移动,到距离平衡位置0.2350 nm时(图4中B点位置)体系到达结合能的一个局部极小值点,Na+(2)与右侧氧原子的作用削弱了原有的O…H—N氢键,相应降低了体系的稳定性;随着Na+(2)继续向β正方向移动,到距离平衡位置0.6150 nm时(图4中C点位置)体系到达结合能的一个局部极大值点;Na+(2)继续向β正方向移动,结合能下降,直到Na+(2)移动到距平衡位置3.1650 nm后结合能约等于0.00 kJ·mol-1, Na+(2)与G-tetrad完全分开.
2.2.3 Na+(2)-G-tetrad中Na+(2)沿γ方向移动

图5 Na+(2)沿γ方向移动的俯视图(a)和结合能变化曲线(b)Fig.5 The vertical view(a)and the binding energy curve(b)of Na+(2)moving along the γ orientationThe data in parentheses denote the coordinates of extremal points for Na+(2)-G-tetrad when Na+(2)moves along the γ orientation.
同样Na+(2)沿γ正负方向的移动也是对称的,以下以γ正方向移动为例进行详细分析.Na+(2)沿γ方向移动指:第1片G-tetrad的坐标不变,只是沿第1片G-tetrad中两鸟嘌呤的对角线在平面I中的投影方向移动,改变Na+(2)到平衡位置的距离,如图5(a)所示.首先将Na+(2)沿γ正方向每隔0.0200 nm取一个结构,到0.8200 nm后每隔0.2000 nm取一个结构,到1.8200 nm后每隔0.5000 nm取一个结构,直到3.8200 nm.图5(b)展示了MP2水平和ABEEMσπ/MM模型下,Na+(2)沿γ方向移动时,Na+(2)-G-tetrad的结合能变化曲线.
与Na+(2)沿β方向移动一致,仍然是平衡位置时(图5中A点位置)结合能最大,体系最稳定.随着Na+(2)向γ正方向移动,体系稳定性降低.到距离平衡位置0.3600和0.5800 nm时(图5中B点和D点位置)体系到达结合能的局部极小值点,到距离平衡位置0.4600和0.8000 nm时(图5中C点和E点位置)体系到达结合能的局部极大值点,直到Na+(2)距离平衡位置2.3200 nm后结合能约等于0.00 kJ· mol-1,Na+(2)与G-tetrad完全分开.
对于 Na+(2)的这三条移动路线,MP2和ABEEMσπ/MM方法计算的结合能的绝对平均偏差分别为7.36、6.07和12.30 kJ·mol-1,表明ABEEMσπ/ MM方法计算的结合能与MP2方法得到的结果有很好的一致性.
2.3 Na+和两片G-tetrad作用的结合能
以上结果说明ABEEMσπ/MM模型能很好地模拟MP2水平上,一个Na+在一片G-tetrad中的移动路线.由于受计算机的限制,从头算很难模拟两片甚至更大的体系.以下应用ABEEMσπ/MM模型预测了G-四链体中Na+的移动路线.

图6 Na+(2)-G-tetrads中Na+(2)分别沿α(a)、β(b)、γ(c)三个方向移动和Na+(3)-G-tetrads中Na+(3)分别沿α(a′)、β′(b′)、γ′(c′)三个方向移动时的结合能变化曲线Fig.6 Binding energy curves of Na+(2)-G-tetrads for Na+(2)moving along the α(a),β(b),γ(c)orientations and Na+(3)-G-tetrads for Na+(3)moving along the α(a′),β′(b′),γ′(c′)orientationsThe data in parentheses denote the coordinates of extremal points along the corresponding orientation.
分别取第1和第2片G-tetrad和其中的Na+(2) (Na+(2)-G-tetrads),以及第2和第3片G-tetrad和其中的Na+(3)(Na+(3)-G-tetrads)作为平衡结构(图2).对于Na+(2)-G-tetrads结构,Na+(2)同样沿α、β、γ三个方向移动.对于Na+(3)-G-tetrads结构,通过Na+(3),且垂直于α方向取一个平面II,将第2片G-tetrad中两鸟嘌呤的夹缝方向在平面II中的投影定义为β′方向;将第2片G-tetrad中两鸟嘌呤的对角线方向在平面II中的投影定义为γ′方向.以Na+(3)所在位置(平衡位置)为起点,讨论了该离子分别沿α、β′、γ′三个方向的移动.由于沿各条路线移动时,正负方向的移动是对称的,所以仅以向正方向移动为例进行详细分析.通过比较各条移动路线中,Na+与两片G-tetrad作用的结合能,来判断Na+的移动路线.结合能计算公式如下:

其中E[Na+]为Na+的能量,E[G-tetrads]为两片G-tetrads复合物的能量,E[Na+-G-tetrads]为Na+-G-tetrads复合物的能量.
图6展示了ABEEMσπ/MM模型下,Na+(2)和Na+(3)沿各条路线移动时,Na+-G-tetrads体系的结合能变化曲线.从图中可以看出,对于α方向,Na+(2)-G-tetrads和Na+(3)-G-tetrads中的Na+都是处于平衡位置(图6中A点)时,体系的结合能最大,结构最稳定.Na+(2)和Na+(3)分别沿α方向移动时,分别需克服564.63和562.10 kJ·mol-1的结合能移出G-四链体.对于β、γ、β′和γ′四个方向,图6各图中B点和D点位置是体系结合能的局部极小值点;C点位置是体系结合能的局部极大值点.其中图6(b)中D点代表Na+(2)与G-tetrads中的N42距离(0.1680 nm)较近,vdW排斥作用(-2856.41 kJ·mol-1)较大;图6 (c)、6(b′)、6(c′)中B点分别代表Na+(2)或Na+(3)与G-tetrads中的O61、N40、O39距离(0.1510、0.1480、0.1190 nm)较近,vdW排斥作用(-1775.25、-2356.86、-31836.73 kJ·mol-1)较大.理论上,存在这么大的排斥能Na+很难从β、γ、β′和γ′四个方向移出.综上所述,可以预测G-四链体中的三个Na+最有可能沿α方向依次移出.
3 结论
Na+-G-四链体是一个明显的极化体系,而ABEEMσπ/MM模型较其它力场能更好地反映体系的极化现象,因此应用ABEEMσπ/MM模型对此体系进行了研究.ABEEMσπ/MM模型验证了:Na+-G-tetrad结构采用平行型的氢键,而Na+不存在时, G-tetrad的四个鸟嘌呤通过交叉型氢键存在.Na+的存在改变了G-tetrad的氢键方式.通过研究一片G-tetrad和一个Na+的相互作用表明:Na+沿α方向移动与平面I的距离为0.1000 nm时,体系最稳定,而实验构型中的K+在平面I内时体系最稳定.并且该方法计算的Na+(2)-G-tetrad体系中Na+(2)分别沿α、β、γ三个方向移动的结果与从头算(MP2)的结果有很好的一致性,充分验证了模型的准确性.在此基础上,应用ABEEMσπ/MM模型研究了Na+(2)-G-tetrads和Na+(3)-G-tetrads中两个Na+的移动路线.由于两个Na+沿β、γ、β′和γ′方向移动时,与两片G-tetrad之间存在很大的排斥能,因此无法从这四个方向移出.而这两个Na+沿α方向移动,需克服的结合能相对较小,由此可以预测,G-四链体中的三个Na+最有可能沿α方向依次移出.本文对Na+-G-tetrad和Na+-G-tetrads体系的研究和探讨,为进一步应用ABEEMσπ/ MM模型进行G-四链体中离子交换通道的动力学模拟打下坚实的基础.
致谢: 感谢Jay William Ponder教授(Department of Biochemistry&Molecular Biophysics,School of Medicine,Washington University)提供Tinker程序.
1 Han,H.Y.;Hurley,L.H.Trends Pharm.Sci.,2000,21:136
2 Sket,P.;Cmugelj,M.;Plavec,J.Bioorg.Med.Chem.,2004,12: 5735
3 Davis,J.T.Angew.Chem.Int.Edit.,2004,43:668
4 Shafer,R.H.;Smirnov,I.Biopolymers,2000,56:209
5 Moine,H.;Mandel,J.L.Science,2001,294:2487
6 Arthanari,H.;Bolton,P.H.Chem.Biol.,2001,8:221
7 Lane,A.N.;Jenkins,T.C.Curr.Org.Chem.,2001,5:845
8 Pennarun,G.;Granotier,C.;Gauthier,L.R.Oncogene,2005,24: 2917
9 Burger,A.M.;Dai,F.;Schultes,C.M.Cancer Res.,2005,65: 1489
10 Tuntiwechapikul,W.;Lee,J.T.;Salazar,M.J.Am.Chem.Soc., 2001,123:5606
11 Cuesta,J.;Read,M.A.;Neidle,S.Min.Rev.Med.Chem.,2003,3: 11
12 Kerwin,S.M.Curr.Pharm.Des.,2000,6:441
13 Neidle,S.;Read,M.A.Biopolymers,2000,56:195
14 Perry,P.J.;Arnold,J.R.P.;Jenkins,T.C.Expert.Opin Investig. Drugs,2001,10:2141
15 Alberti,P.;Lacroix,L.;Guittat,L.;Helene,C.;Mergny,J.L.Min. Rev.Med.Chem.,2003,3:23
16 Hud,N.V.;Smith,F.W.;Anet,F.A.;Feigon,J.Biochemistry, 1996,35:15383
17 Qin,Y.;Hurley,L.H.Biochimie,2008,90:1149
18 Shen,X.Y.;Lü,Y.;Li,S.M.Acta Phys.-Chim.Sin.,2009,25: 783 [沈新媛,吕 洋,李慎敏.物理化学学报,2009,25:783]
19 Meng,F.C.;Xu,W.R.;Liu,C.B.Chem.Phys.Lett.,2004,389: 421
20 Gu,J.;Leszczynski,J.J.Phys.Chem.A,2000,104:6308
21 Ma,L.;Iezzi,M.;Kaucher,M.S.;Lam,Y.F.;Davis,J.T.J.Am. Chem.Soc.,2006,128:15269
22 Tsuzuki,S.;Uchimaru,T.;Matsumura,K.;Mikami,M.;Tanabe,K. J.Chem.Phys.,1999,110:11906
23 Yang,Z.Z.;Wu,Y.;Zhao,D.X.J.Chem.Phys.,2004,120:2541 24 Wu,Y.;Yang,Z.Z.J.Phys.Chem.A,2004,108:7563
25 Yang,Z.Z.;Liu,Y.J.Acta Phys.-Chim.Sin.,2009,25:928 [杨忠志,刘永军.物理化学学报,2009,25:928]
26 Li,X.;Yang,Z.Z.J.Phys.Chem.A,2005,109:4102
27 Qian,P.;Yang,Z.Z.Acta Phys.-Chim.Sin.,2006,22:561 [钱 萍,杨忠志.物理化学学报,2006,22:561]
28 Liu,C.;Yang,Z.Z.Sci.China.Ser.B-Chem.,2009,38:1461 [刘 翠,杨忠志.中国科学B辑:化学,2009,38:1461]
29 Wang,F.F.;Gong,L.D.;Zhao,D.X.J.Mol.Struct.-Theochem, 2009,909:49
30 Li,X.;Yang,Z.Z.J.Chem.Phys.,2005,122:084514
31 Pople,J.A.;Head-Gordon,M.;Raghavachari,K.J.Chem.Phys., 1987,87:5968
32 Parkinson,G.N.;Lee,M.P.H.;Neidle,S.Nature,2002,417:876
September 11,2009;Revised:November 20,2009;Published on Web:December 28,2009.
Mobility of Na+in a G-Quadruplex
GUO Ci LIU Cui YANG Zhong-Zhi*
(School of Chemistry and Chemical Engineering,Liaoning Normal University,Dalian 116029,Liaoning Province,P.R.China)
The Na+-G-quadruplex complex is a polarized system and the mobility of Na+during its formation or decomposition is still unclear.The atom bond electronegativity equalization method at the σπ level fused into molecular mechanics(ABEEMσπ/MM)model clearly defines the lone-pair electron,σ bond and π bond sites in addition to the atomic sites.The partial charge fluctuation is calculated in accordance with a change in the molecular environment and so this method should account well for the polarization effect.In this paper,we discuss some properties for the Na+-G-tetrad complex including its geometry,charge distribution,and binding energy according to the ABEEMσπ/MM method.We also investigate these properties for the Na+-G-tetrad complex using the ab initio method at the MP2/6-31G(d,p)level.The ABEEMσπ/MM results are in good agreement with the ab initio results.The presence of Na+changes the hydrogen bonds in the G-tetrad.By comparing the binding energy of the system for every Na+mobile path, we predict that the most probable path is that three Na+ions move away individually from the G-quadruplex along the α orientation.This study lays a solid foundation for the dynamic simulation of ion exchange channels in a G-quadruplex using the ABEEMσπ/MM model.
G-quadruplex;Na+-G-tetrad;ABEEMσπ/MM method;Ab initio;Mobile path
O641
*Corresponding author.Email:zzyang@lnnu.edu.cn.Tel:+86-411-82159607.
The project was supported by the National Natural Science Foundation of China(20633050,20873055)and Foundation of Department of Education of Liaoning Province,China(2008S133,2009T057,LNET RC0503).
国家自然科学基金(20633050,20873055)和辽宁省教育厅基金(2008S133,2009T057,LNET RC0503)资助项目
