新型2-甲基吲哚衍生物的合成*
2010-11-26何小兰袁玉兵徐海龙王晓宁
何小兰, 袁玉兵, 徐海龙, 王晓宁, 张 焱
(北京服装学院 材料科学与工程学院,北京 100029)
N-取代基-2-甲基-3-甲酰基(乙酰基)吲哚类化合物用途广泛,是合成一些具有光致变色性能的吲哚类俘精酸酐化合物及其衍生物[1,2]的重要中间体。目前国内由于原料试剂的供应有限,研究开发生产较少,许多中间体需要合成。该类化合物的合成,文献主要以2-甲基吲哚、POCl3和N,N-二甲基甲酰胺(DMF)或N,N-二甲基乙酰胺(DMA)为原料,经Vilsmeier-Haack反应合成2-甲基-3-酰基吲哚化合物,再在不同碱的催化下与N-烷基化试剂反应制得[3]。
Vilsmeier-Haack反应是芳香杂环化合物上引入甲酰基或乙酰基的一种应用极为广泛的有机反应,通常用POCl3来生成Vilsmeier-Haack试剂。由于POCl3价格贵且目前国内限制使用,难以获得。本文对其合成工艺进行改进,选用草酰氯替代POCl3,先与DMF或DMA反应生成Vilsmeier-Haack试剂,再与2-甲基吲哚反应,在吲哚的3-位引入甲酰基或乙酰基制得2-甲基-3-甲酰基吲哚(1)或2-甲基-3-乙酰基吲哚(2)。1或2在THF-NaH存在下与烷基化试剂进行烷基化反应,合成了一系列新型的N-取代基-2-甲基-3-甲酰基(乙酰基)吲哚类化合物(3a~3g, 4, Scheme 1),其结构经1H NMR,13C NMR, IR和MS表征。
该方法具有原料价廉易得、反应简单、条件温和、纯化方法简便等优点,适用于工业化生产。本文合成了8个新型吲哚衍生物,为吲哚家族增加了新的成员,为合成吲哚取代的俘精酸酐光致变色化合物奠定了基础。
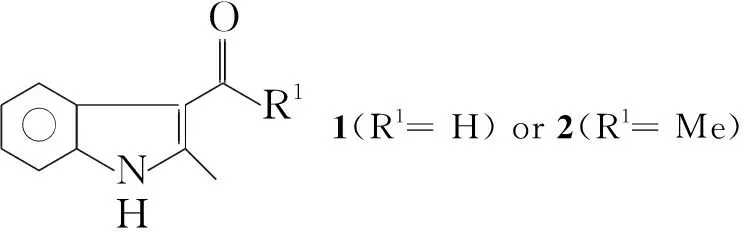
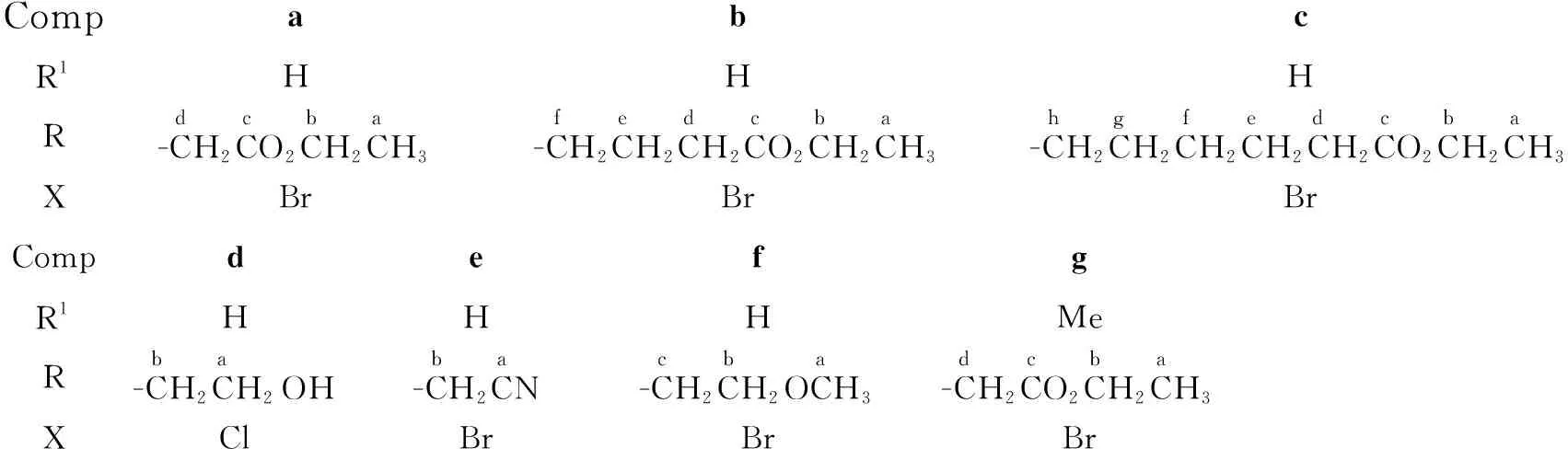
CompabcR1HHHR-CH2CO2CH2CH3dcba-CH2CH2CH2CO2CH2CH3fedcba-CH2CH2CH2CH2CH2CO2CH2CH3hgfedcbaXBrBrBrCompdefgR1HHHMeR-CH2CH2OHba-CH2CNba-CH2CH2OCH3cba-CH2CO2CH2CH3dcbaXClBrBrBr
Scheme1
1 实验部分
1.1 仪器与试剂
XT-4A型电热数字显示熔点仪(温度计未经校正);AvanceⅡ500 MHz型和Avance 300 MHz型核磁共振仪(氘代丙酮为溶剂,TMS为内标);5973 EI-MS型和Esquire 3000 ESI-MS型质谱仪;670 FT-IR型红外光谱仪(KBr压片)。
2-甲基吲哚,Alfa公司;NaH(固含量60%),北京化学试剂公司;THF,化学纯,使用前用钠/二苯甲酮干燥后蒸馏;DMF和DMA,分析纯,用无水硫酸镁干燥过夜后减压蒸馏,收集中间馏分并加入活化的4 Å分子筛储存备用[4];其余试剂均为分析纯。
1.2 合成
(1) 1和2的合成[5]
在三口烧瓶中加入DMF 15 mL,搅拌下于0 ℃~5 ℃缓慢滴加草酰氯0.96 mL(11.27 mmol),滴毕,反应30 min;于室温缓慢滴加2-甲基吲哚0.98 g(7.48 mmol)的DMF(15 mL)溶液,滴毕,反应1 h[TLC监测,展开剂:A=V(石油醚) ∶V(乙酸乙酯)=2 ∶1]。减压蒸除DMF,搅拌下加入冰水(100 mL)中,用饱和碳酸氢钠溶液调至pH 8~9,有大量沉淀生成,过滤,滤饼干燥得白色晶体1 0.75 g,产率63%(若草酰氯用量较大,产率可达96%), m.p.204 ℃(200 ℃~202 ℃[6]);1H NMRδ: 2.77(s, 3H, CH3), 7.16~7.21(m, 2H, ArH), 7.40~7.42(m, 1H, ArH), 8.16~8.18(m, 1H, ArH), 10.19(s, 1H, CHO), 10.91(s, 1H, NH); EI-MSm/z(%): 159[M+, 78], 158{[M-H]+, 100}。
用DMA代替DMF,用类似的方法合成淡黄色固体2,产率35%, m.p.198 ℃~200 ℃(196 ℃~198 ℃[5]);1H NMRδ: 2.57(s, 3H, COCH3), 2.78(s, 3H, 2-CH3), 7.14~7.19(m, 2H, ArH), 7.38~7.42(m, 1H, ArH), 8.10~8.12(m, 1H, ArH), 10.87(s, 1H, NH); EI-MSm/z(%): 173[M+, 48], 158{[M-CH3]+, 100}。
(2) 3的合成(以3a为例)[7]
在三口烧瓶中加入1 1.04 g(6.54 mmoL)和无水THF 20 mL,搅拌5 min使其完全溶解;于0 ℃~5 ℃缓慢加入NaH 0.46 g(11.50 mmoL),有大量气泡冒出,反应30 min。缓慢滴加溴乙酸乙酯0.96 mL(8.68 mmol),滴毕,于室温反应30 min(TLC监测,展开剂:A=1 ∶1)。搅拌下缓慢倒入饱和氯化铵溶液(100 mL)中,用乙酸乙酯(3×50 mL)萃取,合并有机层,用无水MgSO4干燥过夜。减压蒸除乙酸乙酯得N-乙氧基羰甲基-2-甲基-3-甲酰基吲哚(3a)。
用类似的方法合成3b~3g,反应条件和产率见表1。
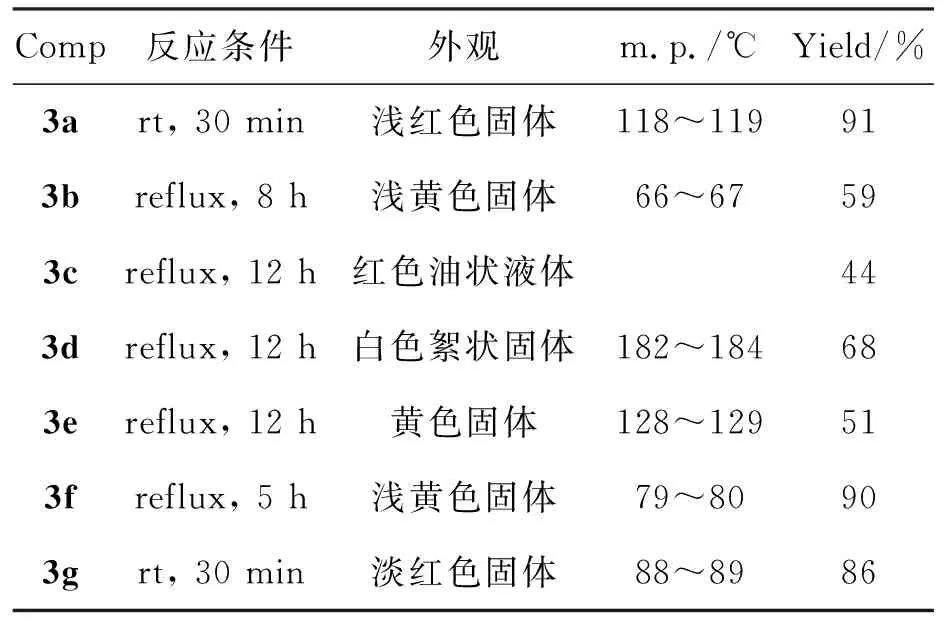
表1 3的合成条件及产率*Table 1 Synthesizing conditions and yield of 3
*其余反应条件同1.2(2)
3a:1H NMRδ: 1.26(t,J=7.0 Hz, 3H, a-H), 2.75(s, 3H, 2-CH3), 4.22(q,J=7.0 Hz, 2H, b-H), 5.18(s, 2H, d-H), 7.23~7.26(m, 2H, ArH), 7.45~7.48(m, 1H, ArH), 8.22~8.24(m, 1H, ArH), 10.24(s, 1H, CHO); IRν: 3 079, 2 942, 2 819, 2 755, 1 737, 1 641, 1 548, 1 513, 1 468, 1 427, 1 379, 1 215, 1 067, 802, 738 cm-1; EI-MSm/z(%): 245[M+, 78], 172{[M-CO2Et]+, 100}。
3b:1H NMRδ: 1.22(t,J=7.5 Hz, 3H, a-H), 2.13(m, 2H, e-H), 2.49(t,J=7.0 Hz, d-H), 2.81(s, 3H, 2-CH3), 4.12(q,J=7.0 Hz, 2H, b-H), 4.35(t,J=7.5 Hz, 2H, f-H), 7.22~7.30(m, 2H, ArH), 7.58(d,J=8.0 Hz, 1H, ArH), 8.23(d,J=7.5 Hz, 1H, ArH), 10.21(s, 1H, CHO);13C NMR(CDCl3)δ: 184.2(3-CHO), 172.5(c-C), 147.1, 136.3, 125.9, 123.2, 122.8, 120.9, 114.4, 109.5(Ar-C), 60.8(b-C), 42.4(f-C), 30.8(d-C), 24.5(e-C), 14.2(a-C), 10.4(2-CH3); IRν: 3 055, 2 992, 2 920, 2 849, 2 809, 2 746, 1 723, 1 644, 1 532, 1 461, 1 446, 1 375, 1 208, 1 053, 1 014, 761 cm-1; EI-MSm/z(%): 273[M+, 100], 172{[M-(CH2)2CO2Et]+, 71}。
3c:1H NMRδ: 1.21(t,J=7.0 Hz, 3H, a-H), 1.48(m, 2H, f-H), 1.67(m, 2H, e-H), 1.87(m, 2H, g-H), 2.31(t,J=7.5 Hz, 2H, d-H), 2.79(s, 3H, 2-CH3), 4.08(q,J=7.0 Hz, 2H, b-H), 4.29(t,J=7.5 Hz, 2H, h-H), 7.21~7.28(m, 2H, ArH), 7.53(d,J=7.5 Hz, 1H, ArH), 8.22(d,J=7.5 Hz, 1H, ArH), 10.20(s, 1H, CHO); IRν: 3 053, 2 994, 2 921, 2 848, 2 810, 2 748, 1 725, 1 643, 1 530, 1 462, 1 448, 1 373, 1 210, 1 016, 860, 759 cm-1; EI-MSm/z(%): 301[M+, 83], 173{[M-CH2=CH(CH2)2CO2Et]+, 100}。
3d:1H NMRδ: 2.81(s, 3H, 2-CH3), 3.96(q,J=5.5 Hz, 2H, a-H), 4.21(t,J=5.5 Hz, 1H, OH), 4.39(t,J=5.0 Hz, 2H, b-H), 7.19~7.26(m, 2H, ArH), 7.51(t,J=7.5 Hz, 1H, ArH), 8.21(t,J=6.5 Hz, 1H, ArH), 10.18(s, 1H, CHO);13C NMR(CDCl3)δ: 183.6(3-CHO), 149.0, 136.7, 125.9, 122.5, 122.0, 120.5, 114.1, 110.0(Ar-C), 60.2(a-C), 45.7(b-C), 9.7(2-CH3); IRν: 3 444, 3 299, 2 823, 1 632, 1 578, 1 531, 1 457, 1 394, 1 372, 1 054, 851, 755 cm-1; EI-MSm/z(%): 203[M+, 100], 172{[M-CH2OH]+, 82}。
3e:1H NMR(CDCl3)δ: 2.80(s, 3H, 2-CH3), 5.01(s, 2H, b-H), 7.34~7.38(m, 3H, ArH), 8.31(m, 1H, ArH), 10.24(s, 1H, CHO);13C NMRδ: 184.5(3-CHO), 146.1, 135.6, 125.6, 124.3, 123.8, 121.4, 115.8, 108.7(Ar-C), 113.4(a-C), 30.9(b-C), 10.4(2-CH3); EI-MSm/z(%): 198[M+, 100], 197{[M-H]+, 62}。
3f:1H NMR(CDCl3)δ: 2.72(s, 3H, 2-CH3), 3.27(s, 3H, a-H), 3.69(t,J=5.7 Hz, 2H, b-H), 4.29(t,J=5.1 Hz, 2H, c-H), 7.25~7.30(m, 3H, ArH), 8.28(m, 1H, ArH), 10.17(s, 1H, CHO);13C NMR(CDCl3)δ: 184.4(3-CHO), 148.6, 136.3, 125.9, 123.0, 122.8, 121.1, 114.4, 109.4(Ar-C), 70.6(b-C), 59.2(a-C), 43.5(c-C), 10.6(2-CH3); IRν: 3 043, 2 981, 2 926, 2 889, 2 829, 2 806, 2 740, 1 640, 1 532, 1 455, 1 428, 1 390, 1 369, 1 117, 1 059, 917, 743 cm-1; EI-MSm/z(%): 217[M+, 93], 172{[M-CH2OCH3]+, 100}, 159{[M-CH2=CHOCH3]+, 89}。
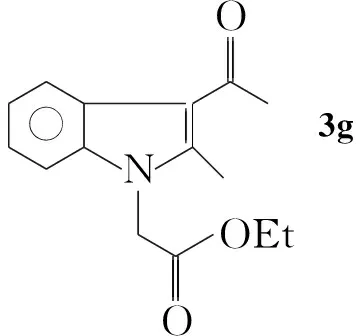
3g:1H NMR(CDCl3)δ: 1.28(t,J=6.0 Hz, 3H, a-H), 2.70(s, 3H, 3-COCH3), 2.75(s, 3H, 2-CH3), 4.24(q,J=6.0 Hz, 2H, b-H), 4.86(s, 2H, d-H), 7.24~7.30(m, 3H, ArH), 7.97~8.01(m, 1H, ArH);13C NMR(CDCl3)δ: 194.8(3-COCH3), 167.5(c-C), 144.7, 136.4, 126.4, 122.4, 122.3, 120.9, 115.0, 109.1(Ar-C), 62.1(b-C), 44.5(d-C), 31.8(3-COCH3), 14.1(a-C), 12.5(2-CH3); IRν: 3 074, 2 941, 2 916, 1 735, 1 640, 1 537, 1 471, 1 436, 1 385, 1 209, 1 064, 783, 732 cm-1; EI-MSm/z: 259[M+, 68], 244{[M-CH3]+, 100}, 216{[M-COCH3]+, 50}。
(3)N-羧甲基-2-甲基-3-乙酰基吲哚(4)的合成
在三口烧瓶中依次加入3g0.50 g(1.93 mmoL)和THF 25 mL,搅拌5 min使其充分溶解。加入30%KOH溶液1.3 mL,回流反应4 h。冷却至室温,用30 mL蒸馏水溶解固体后用2 mol·L-1HCl调pH至3~4。用氯仿(3×30 mL)萃取,合并有机层,旋蒸除氯仿得淡红色固体40.42 g,产率94%, m.p.351 ℃;1H NMR(DMSO-d6)δ: 2.60(s, 3H, COCH3), 2.69(s, 3H, 2-CH3), 4.84(s, 2H, d-H), 7.18(m, 2H, ArH), 7.38(m, 1H, ArH), 7.98(m, 1H, ArH);13C NMR(DMSO-d6) δ: 193.6(3-COCH3), 169.9(c-C), 145.9, 136.9, 126.4, 121.9, 121.8, 120.8, 113.7, 110.6(Ar-C), 46.7(d-C), 31.9(3-COCH3), 12.9(2-CH3); IRν: 3 459, 3 053, 2 967, 1 728, 1 636, 1 513, 1 469, 1 416, 1 383, 1 206, 1 156, 1 116, 972, 892, 739 cm-1; ESI-MSm/z(%): 232{[M+H]+, 100}。
2 结果与讨论
2-甲基吲哚在草酰氯的存在下通过Vilsmeier-Haack反应在3-位引入甲酰基或者乙酰基,得到了2-甲基吲哚的3-位取代衍生物1和2,产率分别为96%和35%。在THF-NaH反应体系中,1或2与一系列烷基化试剂进行烷基化反应,合成了一系列新型的N-取代基-2-甲基-3-甲酰基(乙酰基)吲哚衍生物(3a~3g),产率在44%~91%。3g在氢氧化钾水溶液中水解得到羧酸衍生物4。 1和2的分析结果与文献值一致。3a~3g和4均为未见文献报道的新化合物,1H NMR,13C NMR, IR和MS分析结果证实与Scheme 1结构吻合。
在1或2的合成中,DMF和DMA在使用前必须经除水精制。实验尝试用草酰氯代替POCl3,在吲哚3-位引入甲酰基时,草酰氯过量,既是反应物也是反应溶剂,室温下反应1 h,即可完成反应,产率高达96%;但在吲哚3-位引入乙酰基时,草酰氯大过量,室温下2的产率仅35%。本文选用草酰氯制备Vilsmeier-Haack试剂,可避免加热反应的条件;以草酰氯作反应溶剂,由于其沸点低,易回收,且反应中无副产物产生,大大简化后处理步骤,该实验条件易实现工业化。
在3的合成中,1或2与相应的烷基化试剂进行烷基化反应,较容易得到相应的产物,且产率都较高。只有当烷基化试剂是溴己酸乙酯时,得到的3d的产率只有44%,选用DMF作反应溶剂,可将反应收率提高到57%。
[1] Craig J T, Mason A W, Robert R B,etal. Improved synthesis of indolyl fulgides[J].Journal of Organic Chemistry,2001,66(5):1914-1918.
[2] Yasushi Y. Fulgides for memories and switches[J].Chemical Reviews,2000,100(5):1717-1739.
[3] Liang Y C, Dvornikov A S, Rentzepis P M. Synthesis and properties of photochromic fluorescing 2-indolyl fulgide and fulgimide copolymers[J].Macromolecules,2002,35(25):9377-9382.
[4] 程能林. 溶剂手册(第四版)[M].北京:化学工业出版社,2003.
[5] 李昌秀,柯以侃. 光致变色的吲哚类俘精酸酐(E)-1-苄基-2-甲基-3-吲哚亚乙基丁二酸酐的合成[J].北京化工大学学报,1998,25(1):71-76.
[6] Laureano C, Rodriguez J G, Juan B S,etal. Synthesis,structure and anti-fungal activity of 3-(2′-nitrovinyl) indoles[J].Journal of Medicinal Chemistry,1989,24(1):39-42.
[7] Liang Y C, Dvornikov A S, Rentzepis P M. Photochemistry of photochromic 2-indolylfulgides with substituents at the 1′-position of the indolylmethylene moiety[J].Journal of Photochemistry and Photobiology A:Chemistry,2001,146(1-2):83-93.
