气相色谱法测定锯叶棕片中β-谷甾醇含量
2010-10-20北京市朝阳区药品检验所100101许佳岳福艺
北京市朝阳区药品检验所(100101) 许佳 岳福艺
中国药品生物制品检定所(100050) 孙磊
锯叶棕片系锯叶棕(沙巴棕)Serenoa repens(Bartram) Small成熟的干燥果实提取物制成的天然植物制剂,主治前列腺增生,β-谷甾醇是其有效成分之一。本试验在参考β-谷甾醇相关测定文献[1~3]的基础上,优化建立了GC含量测定方法,为该制剂的质控提供了参考。
1 仪器与试药
Agilent7890A气相色谱仪(FID检测器),β-谷甾醇对照品(中国药品生物制品检定所),甲醇、二氯甲烷、氢氧化钾(分析纯,北京化工厂),实验用水为Milli-Q系统制备。
2 方法与结果
2.1 色谱条件 DB-5毛细管柱(30m×0.25mm×0.25µm);程序升温,初始温度120℃保持2分钟,以30℃·min-1速率升至300℃,保持12分钟;分流比10:1;进样口温度280℃;检测器温度300℃。β-谷甾醇的理论板数为70000,与相邻成分分离度大于1.5。对照品、供试品及溶剂色谱图见附图。
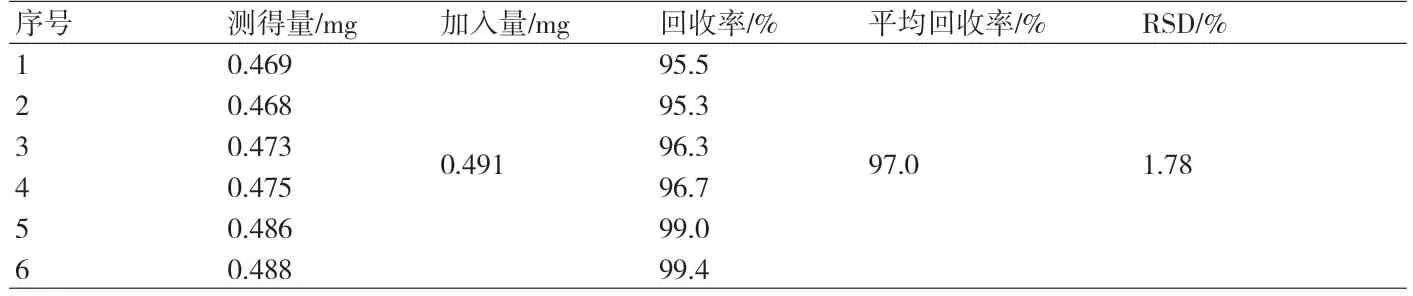
附表 加样回收率结果
2.2 对照品溶液 精密称取β -谷甾醇对照品19.68mg,置10mL量瓶中,加二氯甲烷至刻度,摇匀。
2.3 供试品溶液 取供试品细粉2g置100ml圆底烧瓶中,精密称定,加入13%的甲醇制氢氧化钾溶液20ml(13g 氢氧化钾溶于20ml水中,加甲醇稀释至100ml),回流2小时。放冷,将提取液转移至150ml分液漏斗中,用水30ml洗涤圆底烧瓶,合并洗涤液。提取液用乙醚萃取3次,每次30ml,合并乙醚液。用水洗涤乙醚液3次,每次30ml。乙醚液蒸干,残渣加二氯甲烷溶解并转移至5ml量瓶中。
2.4 线性及范围 精密量取对照品溶液0.2ml、0.5ml、1.0ml、1.5ml、2.0ml分别置10ml量瓶中,加二氯甲烷至刻度,摇匀。精密量取上述溶液各1µl注入气相色谱仪,以进样量(µg)为横坐标,峰面积为纵坐标,绘制标准曲线,得回归方程:y=471.2x-2.5,相关系数r=0.9996,线性范围0.03936µg~0.3936µg。
2.5 精密度 照2.3项下方法制备供试品溶液1份,连续进样6次,β-谷甾醇峰面积RSD为1.62%。
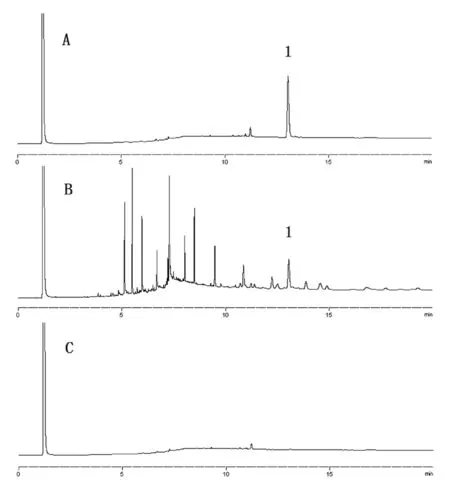
附图 气相色谱图
2.6 稳定性 照2.3项下方法制备供试品溶液1份,在10h内分别进样,β-谷甾醇峰面积RSD为2.83%。
2.7 重复性 照2.3项下方法制备供试品溶液6份,测定,β-谷甾醇含量RSD为2.61%。
2.8 回收率 取供试品细粉1 g,精密称定,置100ml圆底烧瓶中,加入浓度为0.491mg·ml-1的β-谷甾醇对照品溶液1ml,挥干,后续照2.3项下方法操作,结果见附表。
2.9 供试品测定 按上法测定5批供试品,β-谷甾醇含量分别为0.035%、0.034%、0.039%、0.039%和0.038%。
3 讨论
3.1 β-谷甾醇的常用含测方法有薄层扫描法、高效液相色谱法和气相色谱法。其中,薄层扫描法准确度和重现性稍差;高效液相色谱法往往需要使用蒸发光散射检测器才有更好的灵敏度;本试验采用了较简便准确的气相色谱法。
此外,气相色谱法测定β-谷甾醇多进行衍生化,本试验结果表明,不衍生也有较好的灵敏度且方法简单、重现性良好。
3.2 由于β-谷甾醇在二氯甲烷溶解度好,故作为提取溶剂。前处理方法曾试验简单的超声提取方法,但由于本品β-谷甾醇含量低且辅料干扰严重,导致无法准确定量。因此,最终采用了碱液皂化再经乙醚萃取、浓缩的前处理方法。
