抗弓形虫药物研究进展
2010-09-21张瑞岩商立民吴永魁
张瑞岩,刘 全,商立民,吴永魁
(解放军军事医学科学院军事兽医研究所,吉林长春 130062)
弓形虫属于真球虫目肉孢子虫科弓形虫属,是一种专性细胞内寄生原虫。1908年,由两位法国学者在刚地梳趾鼠的肝脾单核细胞内发现。因虫体呈弓形,故命名为刚地弓形虫。人类弓形虫感染率极高,一般为20%~50%,最高达94%。从20世纪80年代我国开始对弓形虫感染情况进行普查,平均感染率约为5.17%。患弓形虫病的孕妇中有30%~46%可能将弓形虫传染给胎儿,当感染发生在妊娠头3个月,多引起流产,也可发生死胎,或生下发育缺陷的无生活能力患儿,其已被列为感染性致畸(TORCH)综合征主要原因之一。对于免疫功能严重损害或抑制者,弓形虫同样是引起致死性病变的主要病原体之一。据报道,在血清学阳性的AIDS患者中,约有30%~40%发生弓形虫脑炎。动物感染弓形虫后,一方面影响其生产性能,另一方面成为人类弓形虫病的传染源,因此,对弓形虫病的防治具有重要的经济学意义和公共卫生学意义。传统的抗弓形虫药物包括乙胺嘧啶、磺胺嘧啶等,然而由于这些药物的毒副作用较大,用药时间长,只能杀死速殖子,停药后易复发,极大的限制了它们的应用。因此,寻找一种高效、低毒的抗弓形虫药物越来越引起人们的重视。
1 干扰叶酸合成类药物
与哺乳动物细胞不同,弓形虫不能直接摄取环境中的叶酸,必须利用对氨苯甲酸在二氢叶酸合成酶的作用下合成二氢叶酸,然后在二氢叶酸还原酶的参与下合成活化型的四氢叶酸。因此,干扰弓形虫的叶酸代谢途径可以有效的抑制和杀灭弓形虫。
1.1 二氢叶酸合成酶颉颃剂类药物
目前用于治疗弓形虫病的二氢叶酸合成酶颉颃剂主要是磺胺类药物,其化学结构与对氨苯甲酸相似,可竞争性的与弓形虫的二氢叶酸合成酶结合,抑制其活性,从而阻碍二氢叶酸的合成,发挥抗弓形虫活性。在体外各种磺胺类药物对弓形虫的半数抑制浓度(IC50)差别很大,从0.6 mg/L(磺胺甲噁唑)到大于30 mg/L(磺胺多辛),其中磺胺嘧啶的IC50为2.5 mg/L。各种磺胺类药物的体内抗弓形虫活性强弱亦存在差异,这与它们的血浆蛋白结合率、维持时间和给药途径等有关。大多数磺胺类药物口服后在小肠上段被迅速吸收,并且与血浆蛋白结合,暂时失去活性,但是这种结合并不牢固,它们能够再分离出来发挥抗弓形虫活性。一般来说,与血浆蛋白结合率高的磺胺类药物排泄较慢,药物半衰期长,例如磺胺多辛的半衰期可长达230 h,而与血浆蛋白结合率低的磺胺类药物在脑脊液中的浓度较高,如磺胺嘧啶的半衰期为17 h,其在脑脊液中的浓度为血液中药物浓度的40%~80%,因此磺胺嘧啶对治疗弓形虫脑炎有特效。磺胺类药物吸收后可分布到全身各组织和体液中,并可通过胎盘屏障进入胎血和羊水中发挥抗虫活性。临床上磺胺类药物并不单独用于治疗和预防弓形虫病,需要与乙胺嘧啶等二氢叶酸还原酶抑制剂联合应用发挥抗弓形虫作用。一般磺胺类药物用药时间较长,其乙酰化代谢产物易引起结晶尿、蛋白尿等造成泌尿系统损伤,有些患者还出现过敏、白细胞减少和溶血性贫血等不良反应,孕妇服用此类药物后会使胎儿产生黄疸样症状,严重者可引起胎儿畸形。
1.2 二氢叶酸还原酶抑制剂类药物
作为二氢叶酸还原酶抑制剂乙胺嘧啶和甲氧苄啶均能阻断弓形虫叶酸代谢途径,发挥抗弓形虫活性。体外试验显示0.05 mg/L的乙胺嘧啶便能够对弓形虫速殖子产生抑制和杀灭作用,而甲氧苄啶在浓度不小于2 mg/L时才能发挥作用。Piketty C等通过口服给予乙胺嘧啶,治疗患急性弓形虫病的老鼠模型,并对老鼠血液、肺和脑内的弓形虫计数,发现乙胺嘧啶对患急性弓形虫病的老鼠具有保护作用,但是停药后出现复发现象。乙胺嘧啶和甲氧苄啶在人体内吸收迅速,分布广泛,半衰期分别为96 h和7.5 h左右,并且均可通过血脑屏障和胎盘屏障。由于乙胺嘧啶对胎儿具有致畸作用,因此在妊娠期的前3个月禁止用于弓形虫病的防治。由于这两种药物对人类二氢叶酸还原酶同样会产生抑制作用,影响胸腺嘧啶核苷酸的合成,所以应用乙胺嘧啶和甲氧苄啶治疗弓形虫病时要适时补充叶酸制剂。Rosow sky A等[1]研究了 2,4-二氨基-5-[2-甲氧-5-(ε-羧基烷氧)苄基]嘧啶类同系物对弓形虫和小鼠肝细胞内二氢叶酸还原酶的抑制作用。结果显示其具有非常高的抑制弓形虫二氢叶酸还原酶活性,然而对小鼠体内的酶也具有一定的抑制作用,同时发现其结构与乙胺嘧啶的一种同系物具有很高的相似性,因此,可为优化此类药物的构效关系提供思路。双氢三嗪类药物同样具有抑制二氢叶酸还原酶的活性,Mui E J等[2]测试了两种新三嗪化合物JPC-2067-B和 JPC-2056的抗弓形虫活性,发现 JPC-2067-B能在体内和体外高效抑制弓形虫生长、繁殖,其在体外的 IC50为 20 nmol/L。JPC-2056为JPC-2067-B的前体药,在体内可经甲基化转变为JPC-2067-B发挥抗弓形虫活性。
1.3 两类药物的联合应用
因为磺胺类药物与二氢叶酸还原酶抑制剂分别在叶酸合成的两个环节上发挥阻碍作用,所以无论是在体内还是在体外他们的联合应用都表现出良好的抗弓形虫活性。研究表明,联合应用磺胺嘧啶和乙胺嘧啶能在24 h内清除患急性弓形虫病的老鼠模型组织内的弓形虫。临床上磺胺类药物与二氢叶酸还原酶抑制剂的联合应用已经成功的用于治疗和预防弓形虫引起的多种疾病,为人类抗弓形虫病发挥了重要作用。
2 大环内酯类药物
许多研究结果显示,大环内酯类药物无论是在体内还是在体外均有较强的抗多种弓形虫活性。这类药物不但能够抑制弓形虫速殖子的生长和繁殖,对弓形虫滋养体和包囊亦具有杀灭作用。乙酰螺旋霉素是最早用于治疗人类弓形虫病的大环内酯类药物,并且目前依然用于孕妇早期弓形虫病的治疗。Chang H R等首先应用尿嘧啶掺入法测试了四种大环内酯类药物(罗红霉素、乙酰螺旋霉素、阿奇霉素和A-56268)的体外抗弓形虫活性,结果发现它们都具有很好的抗弓形虫活性,其IC50分别为54、140、147、246 μ mol/L。Araujo F G 等通过连续10 d每日口服给予阿奇霉素(200 mg/kg)治疗患急性弓形虫病的小鼠,结果阿奇霉素对接种五种不同株弓形虫(其中两株分离自艾滋病患者)的小鼠均起到100%的保护作用。Hutchinson-Mark J等的试验也证实阿奇霉素对包囊内弓形虫缓殖子有杀灭作用,而且与乙胺嘧啶或磺胺嘧啶联合应用表现出很好的协同作用。在临床上阿奇霉素单独使用或与干扰素、复方新诺明等联合应用已经成功的治疗了多例弓形虫病患者。由于阿奇霉素的半衰期较长(40 h),因此其在肝、肺、脑等组织内的浓度远大于在血液中的浓度。与同为大环内酯类药物的阿奇霉素不同,克林霉素的半衰期只有3 h~5 h,因此单独使用时需要每天多次给药才能达到有效的药物作用浓度。鉴于克林霉素半衰期短、单独使用效果不理想和它的低毒副作用,因此,常与乙胺嘧啶等其他抗弓形虫药物联合用于治疗脑和眼弓形虫病。由于各种抗弓形虫药物单独使用都不能达到很好的疗效,因此联合用药仍然是目前治疗弓形虫病的的最好方法。Romand S等通过实验发现罗红霉素即使在亚治疗剂量时其与乙胺嘧啶或磺胺嘧啶联合应用也能够明显减少血液和各组织中的弓形虫虫体数。Djurkovic-Djakovic O等联合应用克林霉素和阿托伐醌治疗Me49株弓形虫感染的小鼠,结果,无论是对急性感染还是慢性感染,两种药物联合应用均能显著提高小鼠存活率和减少脑内包囊数。Julian F等联合应用克拉霉素和乙胺嘧啶对患急性弓形虫脑炎的艾滋病病人进行治疗,发现这两种药物的联合应用与乙胺嘧啶和克林霉素或乙胺嘧啶与磺胺嘧啶联合应用的作用效果相似。
大环内酯类药物抗弓形虫的作用机制目前尚不清楚,研究人员普遍认为与其抗菌作用机制相似,即通过与弓形虫核蛋白体50 s亚基结合,阻断肽链的延长,从而影响蛋白质的合成而发挥抗弓形虫活性。此外,大环内酯类药物在治疗孕妇弓形虫感染时的胎盘通透性和对胎儿的影响,还需要进一步试验观察。
3 其他抗菌药、抗病毒药和抗寄生虫药
对临床上已经成功用于治疗细菌、真菌、病毒和其他寄生虫病的药物,其抗弓形虫活性研究将有可能成为未来治疗弓形虫病的有效途径之一。一方面对这些药物的药理活性、毒副作用和临床应用情况的认识,将大大减少药物研发的时间和经费,另一方面弓形虫感染导致的机体死亡多数是由于一些细菌、病毒等的并发感染,所以将抗菌药、抗病毒药用于抗弓形虫的感染,同时也能预防和治疗其他病原微生物对机体的侵害。
3.1 抗菌药物
科研人员对多种抗细菌和真菌药物的驱弓形虫活性进行了试验研究,发现一些高效、低毒的抗菌药物对弓形虫有很强的抑制和杀灭作用,其中包括喹诺酮类、四环素类和抗真菌抗生素等。Khan A A等对曲伐沙星和它的11种同系物的体外抗弓形虫活性和它们的构效关系进行了研究,结果曲伐沙星的 IC50为 2.93 μ mol/L,其余 11 种同系物的 IC50为0.53 μ mol/L ~14.09 μ mol/L,构效关 系的研究表明,在1,8-萘基环的C-5位或是在氮杂环的C-2位上加一个甲基能使药物活性提高4倍~6倍,在氮杂环的6位上加一个-NH2基团同样会明显提高其抗弓形虫活性。Gozalbes R等应用分子拓扑和虚拟计算技术研究了氟喹诺酮和其他24种喹诺酮类药物的体外抗弓形虫活性,结果发现曲伐沙星、格帕沙星、加替沙星、莫西沙星具有很好的体外抗弓形虫活性,同时发现其活性完全依赖于C-6的氟,C-5的甲基团和C-7的氮杂环能够增强药物活性,而C-3的羧酸基团是活性非必需基团。Sonda S等对抗真菌抗生素金担子素A的体外抗弓形虫活性进行了研究,结果,在体外金担子素A能够阻碍弓形虫复制,并使其内部结构发生变化如形成空泡。另外,他们发现金担子素A能够抑制弓形虫的鞘脂合成,而其对哺乳动物细胞的新陈代谢无影响,因此,弓形虫的鞘脂合成途径也被认为是抗弓形虫药物设计的可能靶点。
3.2 抗病毒药物和其他抗寄生虫药物
一些抗病毒药物同样具有抗弓形虫活性,如利托那韦、那非那韦和双脱氧肌苷等。Derouin F等报道利托那韦和那非那韦在低浓度下就能高效抑制弓形虫增长,它们的 IC50分别为 5.4 mg/mL和4.0 mg/mL,同时发现弓形虫具有天冬氨酰蛋白酶,它在复制过程中发挥重要作用。
阿托伐醌和青蒿素类药物已经成功用于治疗和预防疟原虫引起的疾病,其中阿托伐醌在很低浓度时对弓形虫速殖子和缓殖子即具有很好的杀灭作用,对患急性弓形虫病和脑弓形虫病的老鼠能起到很好的保护作用,药物作用部位是弓形虫线粒体的细胞色素b。令人关注的是阿托伐醌对弓形虫包囊同样具有很好的抵抗活性,其可明显减少老鼠体内弓形虫包囊数。但是,有研究显示阿托伐醌对不同株弓形虫的作用效果存在很大差异,其原因尚不完全清楚,有可能是某些虫株对阿托伐醌具有天然抵抗力或是用药后出现了药物抗性突变株。阿托伐醌与磺胺嘧啶和大环内酯类药物等合用有很好的协同作用,临床上已经用于治疗脑、眼弓形虫病。在体外青蒿素及其衍生物能在弓形虫生长繁殖的多个环节发挥抑制作用,同时降低其对宿主细胞的侵染力,并表现出很高的杀灭活性,体内试验显示青蒿素类药物能显著杀灭弓形虫包囊,延长接虫小鼠的存活时间[3-4]。孙志伟等发现双氢青蒿素联合磺胺嘧啶钠治疗小鼠急性弓形虫感染有很好的协同作用,而且可有效防止停药后复发。目前青蒿素类药物的抗弓形虫作用机制仍然没有统一的定论,研究结果展示青蒿素类药物能干扰弓形虫叶酸代谢、影响虫体钙依赖性蛋白分泌、破坏其细胞结构和增强机体免疫力等[5-6]。治疗弓形虫病的关键和难点在于杀灭和清除患者体内的弓形虫包囊,防止其复发,实现彻底治愈。因此,阿托伐醌和青蒿素类药物的抗弓形虫包囊活性受到研究人员的重视,其中阿托伐醌对弓形虫包囊的长期耐药性以及青蒿素类药物杀灭弓形虫包囊的作用机制和药物构效关系将成为未来研究的重点。
由于一些细胞内寄生原虫与弓形虫具有很多相似的生化路径和生理特性,所以用于治疗这些寄生虫病的药物非常值得关注和研究。特别是对同为顶端复合门包子纲的疟原虫的大量研究将给抗弓形虫药物的研究提供重要的理论依据和药物靶点。而对弓形虫和抗弓形虫药物的研究也会为人类对疟疾病的预防和治疗提供新的方法。
4 其他药物
2008年,美国微生物学家Nagamune K等[7]对弓形虫与植物中的生化反应路径进行了比较,结果发现二者存在很多共同的特点,其中一种在植物体内控制应激响应和休眠的荷尔蒙-脱落酸(ABA)引人关注。在植物体内ABA能够诱导第二信使环状腺苷二磷酸核糖(cADPR)的产生,而cADPR控制着细胞内钙的释放,在弓形虫体内cADPR同样控制着细胞内钙的释放。加入ABA能促使弓形虫产生cADPR,刺激钙依赖性蛋白分泌,并且诱导弓形虫从宿主细胞内释出。应用一种普通的除草剂阻断弓形虫制造脱落酸后发现即使弓形虫在宿主细胞内已经繁殖了足够的数量,其仍然不会释出而保持一种无活动的停滞状态。进一步试验研究显示这种除草剂能够挽救被弓形虫感染的小鼠。这一研究成果不但使人们对这种细胞内寄生原虫有了新的认识,而且由于发现其与植物生化路径的相似性,为今后抗弓形虫药物的研究提供了全新的思路和选择。许多试验研究揭示抑制弓形虫体内各种酶的活性能有效的抑制和杀灭弓形虫,如二膦酸盐化合物能通过抑制法尼焦磷酸合酶阻止弓形虫繁殖,其中不含氮的二膦酸盐表现出很高的药物活性(IC50为5 μ mol/L ~ 10 μ mol/L)[8]。Wei S 等[9]报道人类p38促分裂原活化蛋白激酶抑制剂同样可以阻止细胞内弓形虫复制并能够对致死性弓形虫感染的小鼠起到保护作用。最近法国学者发现一种弓形虫组蛋白脱乙酰基酶抑制剂具有很好的抗弓形虫活性,该制剂能诱导弓形虫从速殖子转化为缓殖子,能影响弓形虫大约370个基因的表达,并证实弓形虫组蛋白脱乙酰基酶是弓形虫基因表达和阶段转换的重要调控子[10]。除此之外,一些他汀类药物、维生素D、细胞活素类物质如 IFN-γ、TNF-α、IL-1-α、IL-6 和抗菌肽等均表现出很好的抗弓形虫活性,并且与其他抗弓形虫药物合用具有很好的协同作用。
5 药物靶点
确定药物靶点对研发新药和寻找先导化合物具有重要意义,而且有助于了解药物作用机制,为优化药物设计、提高研发效率奠定基础。迄今为止,对弓形虫的药物靶标不甚明了,所以抗弓形虫药物研究进展缓慢。弓形虫病对人兽危害严重,传统的干扰弓形虫叶酸合成类药物虽然发挥了重要作用,但这些药物不能完全治愈,而且毒副作用大,因此,寻找新的特异性药物靶标已经迫在眉睫。近年来随着分子生物学的发展和人们对弓形虫病研究的不断深入,一些新的潜在药物靶点不断发现(表1),其中弓形虫体内脂质合成途径和一些特异性酶类值得关注,同时,弓形虫体内的独特生理结构(如尖形顶端)和生化反应途径等也具有重要的研究价值。
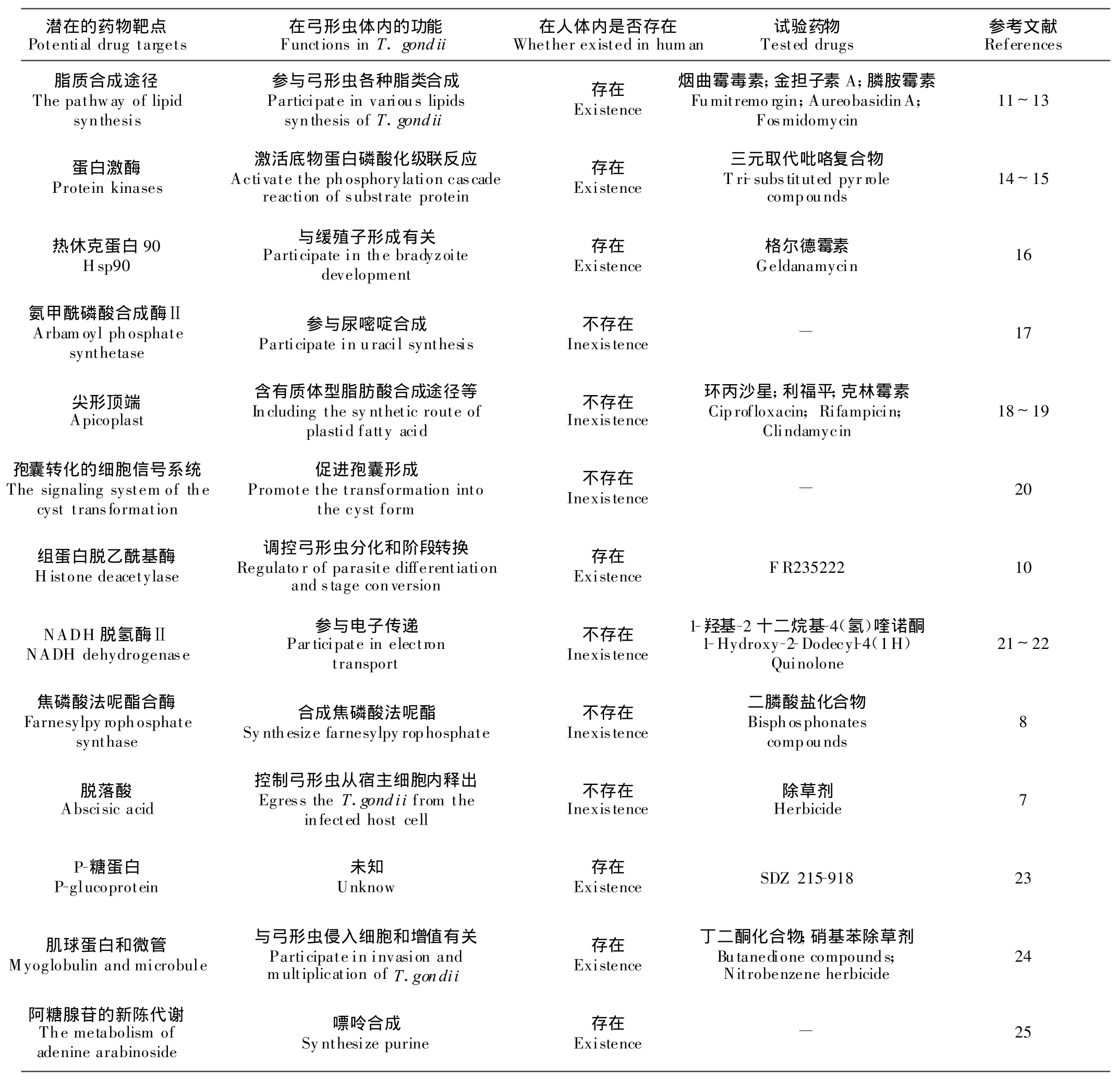
表1 弓形虫体内潜在的药物靶点Table 1 Potential drug targets in Toxoplasma gondii
6 前景和展望
弓形虫在药物或免疫系统的作用下可由速殖子转化成缓殖子,并形成包囊以抵御药物攻击。而在停药后或机体免疫力低下时,可由缓殖子转化成速殖子并迅速繁殖,引起机体病变。由于一般药物只能杀灭速殖子而对包囊内的缓殖子无作用,所以弓形虫病很容易复发,难以完全治愈。因此,发现一种活性高、毒性低、能完全杀灭弓形虫缓殖子、清除包囊的药物将是今后抗弓形虫药物研究的重点和难点。近年来,人们应用现代分子生物学技术对弓形虫生理生化特性进行了大量和深入的研究,特别是针对弓形虫体内独有结构的研究,为新药研发提供理论和实验依据。另外,一些特异的生化反应路径和酶类将成为今后抗弓形虫药物研究的重要资源。而一些抗病原微生物尤其是抗寄生虫新药的发现及临床上的联合用药,将为今后弓形虫病的防治提供重要的思路和方法。
[1]Rosowsky A,Forsch R A,Queener S F,et al.Inhibition of Pneumocystis carinii,Toxoplasma gondii,and Mycobacterium avium dihydrofolate reductases by 2,4-diamino-5-[2-methoxy-5-(omega-carboxyalkyloxy)benzyl]pyrimidines:marked improvement in potency relative to trimethoprim and species selectivity relative to piritrexim[J].J Med Chem,2002,45(1):233-241.
[2]Mui E J,Schiehser G A,Milhous W K,et al.Novel triazine JPC-2067-B inhibits Toxoplasma gondii in vitro and in vivo[J].Plos Negl T rop Dis,2008,2(3):190-202.
[3]D'Angelo J G,Bordón C,Posner G H,et al.A rtemisinin derivatives inhibit Toxoplasma gondii in vitro at multiple steps in the lytic cycle[J].J Antimicrob Chemother,2009,63(1):146-150.
[4]de Oliveira T C,Silva D A,Rostkowska C,et al.Toxoplasma gondii:effects of Artemisia annua L.on susceptibility to infection in ex perimental models in vitro and in v ivo[J].Exp Parasitol,2009,122(3):233-241.
[5]杨翠萍,万红娇.青蒿素及其衍生物抗弓形虫的研究进展[J].国际医学寄生虫病杂志,2006,33(3):160-162.
[6]Nagamune K,Beatty W L,Sibley L D.A rtemisinin induces calcium-dependent protein secretion in the protozoan parasite Toxoplasma gondii[J].Eukaryotic Cell,2007,6(11):2147-2156.
[7]Nagamune K,Hicks L M,Fux B,et al.Abscisic acid controls calcium-dependent egress and development in Toxoplasma gondii[J].Nature,2008,451(7175):207-210.
[8]Szajnman S H,Garc í a Li~nares G E,Li Z H,et al.Synthesis and biological evaluation of 2-alkylaminoethy l-1,1-bisphosphonic acids againstTrypanosoma cruziand Toxoplasma gondii targeting farnesyl diphosphate synthase[J].Bioorg Med Chem,2008,16(6):3283-3290.
[9]Wei S,Daniel B J,Brumlik M J,et al.Drugs designed to inhibit human p38 mitogen-activated protein kinase activation treat Toxoplasma gondii and Encephalitozoon cuniculi infection[J].Antimicrob Agent and Chemother,2007,51(12):4324-4328.
[10]Bougdour A,Maubon D,Baldacci P,et al.Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites[J].J Exp Med,2009,206(4):953-966.
[11]Sonda S,Hehl A B.Lipid biology of Apicomplexa:perspectives for new drug targets,particularly for Toxoplasma gondii[J].Trends Parasitol,2006,22(1):41-47.
[12]Seeber F.Biosynthetic pathways of plastid-derived organellesas potential drug targets against parasitic Apicomplexa[J].Curr Drug Targets Immune Endocr Metabol Disord,2003,3(2):99-109.
[13]Gornicki P.Apicoplast fatty acid biosynthesis as a target for medical intervention in Apicomplexan parasites[J].Int J Parasitol,2003,33(9):885-896.
[14]Doerig C.Protein kinases as targets for anti-parasitic chemotherapy[J].Biochim Biophys Acta,2004,1697(1-2):155-168.
[15]Gurnett A M,Liberator P A,Dulski P M,et al.Purification and molecular characterization of cGMP-dependent protein kinase from Apicomplexan parasites.A novel chemotherapeutic target[J].J Biol Chem,2002,277(18):15913-15922.
[16]Echeverria P C,Matrajt M,Harb O S,et al.Toxoplasma gondii Hsp90 is a potential drug target whose ex pression and subcellular localization are developmentally regulated[J].J M ol Biol,2005,350(4):723-734.
[17]Fox B A,Ristuccia J G,Bzik D J.Genetic identification of essential indels and domains in carbamoyl phosphate synthetase II of Toxoplasma gondii[J].Int J Parasitol,2009,39(5):533 539.
[18]Roos D S.The apicoplast as a potential therapeutic target in Toxoplasma and other apicomplexan parasites:some additional thoughts[J].Parasitol T oday,1999,15(1):5-7.
[19]M cFadden G I,Roos D S.Apicomplexan plastids as drug targets[J].Trends Microbiol,1999,7(8):328-332.
[20]Narasimhan J,Joyce B R,Naguleswaran A,et al.T ranslation regulation by eukary otic initiation factor-2 kinases in the development of latent cysts in Toxoplasma gondii[J].J Bio Chem,2008,283(24):16591-16601.
[21]Lin S S,Kerscher S,Saleh A,et al.The Toxoplasma gondii type-II NADH dehydrogenase T gNDH2-I is inhibited by 1-hydroxy-2-alky l-4(1H)quinolones[J].Biochim Biophys Acta,2008,1777(11):1455-1462.
[22]Saleh A,Friesen J,Baumeister S,et al.Growth inhibition of Toxoplasma gondii and Plasmodium f alciparum by nanomolar concentrations of 1-Hydroxy-2-Dodecy l-4(1H)Quinolone,a high-affinity inhibitor of alternative(Ty pe II)NADH dehydrogenases[J].Antimicrob Agents Chemother,2007,51(4):1217-1222.
[23]Silverman J A,Hayes M L,Luft B J,et al.Characterization of anti-Toxoplasmaactivity of SDZ 215-918,a cyclosporin derivative lacking immunosuppressive and peptidyl-prolylisomerase-inhibiting activity:possible role of a P glycoprotein in Toxoplasma phy siology[J].Antimicrob Agents Chemother,1997,41(9):1859-1866.
[24]chwartzman J,Heintzelman M,Stokkermans T.The cytoskeleton of Toxoplasma gondii as a possible drug target[C].Keyst Symp Mol Cell Biol,1998,108:2-8.
[25]Adenosine metabolism in Toxoplasma gondii:potential targets for chemotherapy[J].Curr Pharma Des,2007,13(6):581-597.
