STK33基因在非小细胞肺癌中表达的临床研究
2010-09-20吕东津许哲源王平彭浩彭俊熊健
吕东津 许哲源 王平 彭浩 彭俊 熊健
昆明医学院附属昆华医院,云南省第一人民医院胸外科,云南 昆明650032
肺癌目前是世界范围内发生率和病死率最高的恶性肿瘤[1],其中85%都是非小细胞肺癌(non-small cell lung cancer,NSCLC)。研究证实EGFR通路在肿瘤(其中就包括了NSCLC)的发生发展中起着至关重要的作用。进入21世纪以来,以EGFR为靶标的分子靶向药物的出现为肺癌的治疗带来了新的希望和手段,以吉非替尼(Gefitinib,ZD1839,易瑞沙,阿斯利康公司)和厄洛替尼(Erlotinib,OSI-774,特罗凯,罗氏公司)为代表的酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI)是目前被多国批准并被广泛应用于进展或难治性NSCLC的小分子靶向药物[2]。但是随着时间的推移,研究发现K-ras基因突变与肺癌患者对TKI药物的原发耐药有关。大量的临床试验也证明了与K-ras未突变患者相比,K-ras突变患者对EGFR抑制剂不太可能有明显的疗效[3-5]。最近Scholl等[6]研究发现与突变K-ras基因依赖的相关肿瘤细胞系中,通过抑制STK33能产生综合致死效应,而对于那些非K-ras依赖的细胞系却没有作用。因此,推测通过抑制STK33这一新思路能巧妙地解决部分NSCLC患者TKI耐药性的困境。
STK33,一种新的丝氨酸/苏氨酸激酶基因(a novel serine/threonine kinase gene,STK33),定位于人类染色体 11p15.3区域[7-8],尽管经过与其他激酶的序列比较,其被确定为钙调节蛋白家族(CAMK)的一员[9-11],但是,目前对于STK33的生化特性和生物功能都不是很清楚。本研究采用Real-time qPCR法及Western blot蛋白印迹法对24例经病理确诊的NSCLC病例中的癌组织、远癌组织中及9例肺良性病变的STK33表达情况进行检测,探讨STK33在NSCLC中的表达规律,及其在NSCLC发生、发展中的临床意义。
1 资料和方法
1.1 资料来源 收集2009年4—9月在云南省第一人民医院心胸外科接受诊断和治疗的NSCLC肺癌患者24例。所有患者均经手术病理确诊,且资料完整、术前未接受任何相关放化疗。其中男性14例,女性10例;年龄范围39~74岁,平均年龄(57±5)岁。对照组采用同一患者的远癌组织(距肿瘤活检切除边缘外3~6 cm的组织)和9例肺良性病变组织,其中炎性假瘤5例,结核2例,淋巴网状组织非典型增生1例,良性肿瘤1例。
1.2 仪器和试剂 电泳设备、凝胶成像系统(Bio Rad公司),7500 fsat PCR仪器(ABI公司)。高纯总RNA快速提取试剂盒、氯仿(Bioteke公司),dNTP Mix(上海生工),Jump Start Taq DNA Polymerase(Sigma公司),SYBR GREENⅠ染液(Amresco Inc),Ribo nuclease Inhibitor、DNaseⅠ(RNase-free)、M-MLV逆转录酶(promega)、100 bp DNA Ladder(Tiangen公司),引物有看家基因β-actin、目的基因STK33(上海生工公司合成)。抗actin一抗、抗STK33一抗(Abcam公司),BCA蛋白浓度测定试剂盒(碧云天生物技术研究所),PMSF(上海生工公司),HRP标记羊抗兔IgG二抗(鼎国生物公司),化学发光检测底物试剂盒(Pierce公司),胶片(柯达有限责任公司),PVDF膜(Millipore 公司)。
1.3 实验方法
1.3.1 组织总RNA的抽提 ⑴匀浆处理:提前把1 mL试剂盒中配的裂解液加入到用1‰DEPC处理过的1.5 mL离心管中,从液氮灌中取米粒大小组织装入冰冻的研钵,并加入少许液氮到研钵中,尽量研磨组织使其成冰冻的粉末状,保持液氮不完全蒸发,并用移液枪挑入1.5 mL离心管,同时轻轻吹打混匀。⑵将匀浆样品剧烈震荡混匀,在15~30 ℃条件下温育5 min以使核蛋白完全分解。⑶4 ℃的条件下12 000 r/min离心10 min,小心取上清液转入一个新的RNase free的离心管中。⑷每1 mL裂解液加0.2 mL氯仿。盖紧样品管盖,剧烈震荡15 s并将其在室温下温育3 min。⑸于4 ℃12 000 r/mim离心10 min,样品会分成3层:下层为有机相,中间层和上层为无色的水相,RNA存在于水相中,水相层的容量大约为所加裂解液体积的60%,把水相转移到新管中,进行下一步操作。⑹加入1倍体积70%乙醇,颠倒混匀,得到的溶液和可能沉淀一起转入吸附柱中。⑺10 000 r/min 离心45 s,弃掉废液,将吸附柱重新套回收集管。⑻加500 μL 去蛋白液,12 000 r/min 离心45 s,弃掉废液。⑼加入700 μL 漂洗液,12 000 r/min 离心60 s,弃掉废液。⑽加入500 μL漂洗液,12 000 r/min离心60 s,弃掉废液。⑾将吸附柱放回空收集管中,12 000 r/min 离心2 min,尽量除去漂洗液,以免漂洗液中残留乙醇抑制下游反应。⑿取出吸附柱,放入1个无RNA水解酶的离心管中,根据预期RNA 产量在吸附膜的中间部位加入50~80 μL无RNA水解酶水解,于室温下放置2 min,12 000 r/min 离心1 min。
1.3.2 测A值定量RNA浓度和电泳检测RNA完整性 ⑴紫外分光光度计进行RNA定量:在比色杯中加98 μL DEPC处理过的双蒸水,再加入2 μL样品,测定A260/A280值,计算:RNA浓度(μg/μL)=A×0.33×稀释倍数(50)。⑵分析RNA样品的纯度:1.9≤A260/A280≤2.1为纯RNA,A260/A280≥2.0为无盐污染。⑶RNA完整性检测:被提取后的RNA的完整性通过凝胶电泳来证实。1.2%的琼脂糖凝胶,以0.5 μg/mL溴化乙锭染色。取1 μL RNA,加入9 μL DEPC H2O及1 μL上样缓冲液,混匀后上样,电泳(60 V,0.5×TBE)40 min,凝胶成像仪检测。
1.3.3 消化基因组DNA 将42.3 μL RNA(<25μg)、5 μL 10×buffer、2 μL DNase Ⅰ(5 U/μL)、0.7 μL RNase抑制剂(40 U/μL)混合,于37 ℃消化30 min,加入2 μL 0.5 mol/L EDTA 72 ℃,10 min灭活DNaseⅠ。
1.3.4 逆转录cDNA合成 ⑴取≤4.0 μg的总RNA,分别加入2.5 μL 6base随机引物(100 μmol)和2.5 μL 9base随机引物(100 μmol),和/或0.5 μL oligo-dT(100 μmol),充分混匀之后,70 ℃,6 min使RNA二级结构延展,然后立即置于冰上2 min以上,使随机引物和/或oligo-dT引物和RNA模板充分结合。⑵向上述混合体系中加入10 μL 5×的逆转录buffer,加入2.5 μL dNTP(10 mmol/L),加入400 U的逆转录酶,加入30 U的RNase抑制剂后置于37 ℃,90 min以上,完成逆转录。
1.3.5 实时荧光定量PCR
1.3.5.1 荧光定量PCR 体系 10×PCR Buffer 2.5 μL,dNTP 0.5 μL(10 mmol/L),正向引物:0.5 μL(10 pmol/μL),反向引物:0.5 μL(10 pmol/μL),20× SYBR GREENⅠ 0.5 μL(终浓度为0.2倍),模板:DNA 1 μL,Jumpstart Taq DNA聚合物0.4 μL(1.25 U),加入dH2O(灭菌蒸馏水)至25.0 μL。
1.3.5.2 实时定量PCR引物 β-actin序列,F:5’-CGGGAAATCGTGCGTGAC-3’;R:5’-CAGGAAGGAAGGCTGGAAG-3’。STK33序列,F:5’-GGTGGATCATTCAAAGTCTCGC-3’;R:5’-GCTAAGCCAAAATCAGTCACCTT-3’。
1.3.5.3 循环条件 95 ℃预变性30 s, 95 ℃变性10 s,60 ℃退火延伸共30 s,扩增共40个循环。每个样本平行检测3次,PCR产物于2%的琼脂糖凝胶电泳检测。β-actin作为内参, H2O作为PCR的阴性对照,检测样品单一的溶解曲线峰及琼脂糖凝胶电泳单一条带为特异性扩增。
1.3.5.4 实时荧光定量RT-PCR产物鉴定及定量分析 ⑴产物鉴定:包括熔解曲线分析和2%琼脂糖凝胶电泳。3个平行样本的熔解曲线只产生一个熔解峰即表明产物是唯一的,结合2%琼脂糖凝胶电泳可进一步鉴定产物的特异性,以DNA分子量标记物为参照,可证明产物片断大小与实验设计相符。⑵通过下列公式定量分析:
△CT(sample)=CT target gene-CT reference gene
△CT(calibrator)=CT target gene-CT reference gene
CT值(cycle threshold)代表循环阈值,指PCR扩增过程中,目的基因cDNA扩增产物荧光信号超过基线值(进入指数增长期)时的循环数,目的基因起始拷贝数越多,达到指数扩增所需循环数越少,即CT值越小。将各样本的CT值与该样本扩增β-actin时的CT值代入公式,即得到各样本STK33的相对表达量,用△CT值表示。(注:△CT值越小表示该基因在组织中的表达越高)。Sample(样本):肺癌患者的癌组织;Calibrator(对照):远癌组织和良性病变患者的肺组织;Target gene(靶基因):STK33;Reference gene(参照基因):β-actin。
1.3.6 Western blot蛋白印迹
1.3.6.1 肺组织总蛋白提取 ⑴取米粒大小标本肺组织,置于液氮预冷的研钵中,在液氮中尽量研碎。⑵弃上清液后用PBS重复洗涤一次。用枪吸干上清液后,向预冷粉末状标本肺组织中加入适量RIPA裂解液,其主要成分为50 mmol/L Tris (pH=7.4),150 mmol/L NaCl,1% NP-40,0.5%脱氧胆酸钠,0.1% SDS,以及EDTA。冰上裂解30 min,裂解过程中要以漩涡震荡器震荡,以使肺组织充分裂解。⑶4 ℃、12 000 r/min离心15~20 min,取上清液分装于0.5 mL离心管中并置于-20 ℃保存。⑷取出蛋白裂解液,用BCA蛋白浓度测定试剂盒蛋白浓度,加入5×还原型上样缓冲液,沸水煮沸10 min。
1.3.6.2 SDS-PAGE电泳 ⑴制备分离胶:分离胶的高度要合适,要保证蛋白在压缩胶里的行程不少于1 cm。当把丙烯酰胺分离胶倒铸之后,用去离子水封闭分离胶,以达到使分离胶水平的目的,同时可以防止空气对于丙烯酰胺的聚合抑制。⑵待分离胶凝聚,除去分离胶表面的去离子水,然后制备压缩胶。⑶待到压缩胶凝聚之后,在电泳槽中加入电泳缓冲液。拔取制胶梳子。⑷对于每个上样孔,上样20 μg或100 μg总蛋白,压缩胶阶段,以60 V电压进行电泳。待溴芬兰条带(即蛋白条带)过了压缩胶和分离胶的分界线之后,可以加高电压以缩短电泳时间。但是,过高的电压会导致发热加快,同时,也会使电泳条带变得不规则。⑸根据marker的位置,决定电泳的时间。待到合适时间,结束电泳。取出分离胶部分进行考马斯R250染色,或者进行Western blot的蛋白转印。⑹在蛋白电泳结束前约半小时,将PVDF膜用甲醇完全浸湿至半透明状,然后用去离子水漂洗2 min。将漂洗过的PVDF膜放置于转移缓冲液中,浸泡最少10 min以上。⑺在浸泡PVDF膜的同时,将上下两层滤纸都进行浸泡。⑻将电泳结束后的分离胶放置于转移缓冲液中,平衡10~15 min。⑼上下两层滤纸与分离胶还有PVDF膜的大小规定如下:下层滤纸=PVDF膜=凝胶>上层滤纸。⑽待到凝胶平衡时间足够后,将下层滤纸放置于转印装置的正极,用玻棒撵出滤纸中的气泡。置PVDF膜于下层滤纸上,玻棒适度挤压。放置分离胶于PVDF膜上,玻棒适当挤压,切忌把分离胶压破,放置上层滤纸于凝胶之上,玻璃棒轻轻挤压出气泡。之后,确定滤纸及胶层和膜之间没有气泡。加上转印装置的负极。以恒流(50 mA)或者恒压(20 V)进行蛋白转印,转印的强度与时间为1 mA/cm2不超过1.5 h。⑾待转印完毕,取出凝胶和PVDF膜。将凝胶进行考马斯亮兰R250染色,检测蛋白转印完全与否。将PVDF膜在洗脱液0.2% Tween 20 in trisbuffered saline (TBST)中漂洗,4×10 min。⑿待PVDF膜漂洗完毕之后,加入封闭液进行封闭,4 ℃过夜。
1.3.6.3 免疫显色 ⑴将封闭过的PVDF膜进行漂洗,4×10 min。⑵用封闭液稀释抗STK33一抗或抗β-Actin一抗,将漂洗过的PVDF膜放置于其中温育,室温(当室温低于25 ℃时,于37 ℃)温育1.5 h。⑶将一抗反应后的PVDF膜进行漂洗,4×10 min。⑷用封闭液稀释二抗(HRP标记羊抗兔IgG二抗),将漂洗过的PVDF膜放置于其中孵育,室温(当室温低于25 ℃时,于37 ℃)温育40 min。⑸将二抗反应后的PVDF膜进行漂洗,4×10 min。⑹以下步骤在暗室中进行,将底物反应液混合配制好,将PVDF膜放置于内,反应10~20 min。⑺将PVDF膜放置暗盒中,压紧暗盒,曝光时间5~10 min。⑻打开暗盒,将经过曝光的胶片在显影液中轻柔漂洗,当目的条带出现之后,立即用清水冲洗,将胶片放置于定影液中漂洗1 min。所有胶片都经过扫描,采用Image J软件进行定量。
1.4 统计处理 所有数据建立Excel数据库,应用SPSS 11.5统计软件进行处理。计量资料用±s 表示;均数比较采用t检验及单因素方差分析;以P<0.05为差异有统计学意义。
2 结 果
2.1 实时荧光定量PCR 和 WB结果
2.1.1 总RNA纯度和完整性鉴定 ⑴实验中所抽提总RNA A260/A280值均在1.74~2.05之间,表明纯度较高。⑵1.2%琼脂糖凝胶电泳,EB染色可见28 S和18 S两条主带清晰,比值接近为2∶1,无拖尾降解现象发生。表明总RNA完整性较好,基本无降解(图1)。
2.1.2 PCR产物鉴定 2%琼脂糖凝胶电泳:将实时荧光定量PCR产物进行2%琼脂糖凝胶电泳,进一步鉴定产物的特异性及验证产物片段大小。β-actin和STK33扩增产物片段长度分别是181和160 bp(图2)。
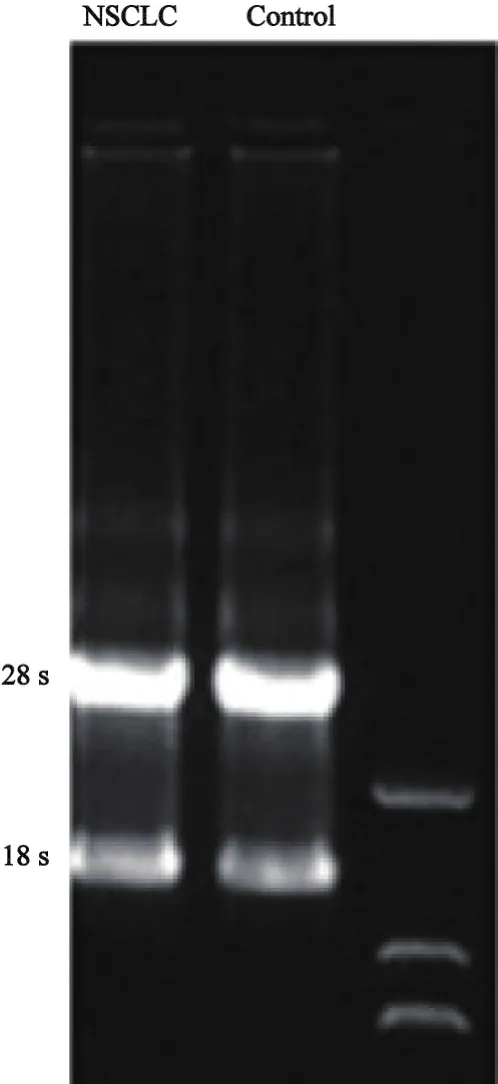
图1 NSCLC组和对照组组织总RNA凝胶电泳结果Fig.1 Agarose gel eletrophoretogram of the total RNA in NSCLC group and control group
2.1.3 实时荧光定量PCR检测STK33 mRNA表达情况的结果分析
2.1.3.1 NSCLC患者肺癌组织和远癌组织中STK33 mRNA的表达情况 实时荧光定量PCR结果显示,NSCLC患者肺癌组织、远癌组织中STK33平均ΔCT值分别为8.7±2.0,9.7±1.4。NSCLC患者肺癌组织STK33基因表达量高于远癌组织,且两者比较差异有统计学意义。(P<0.05,表 1)。
表1 肺癌患者肺癌组织和远癌组织中STK33水平Tab.1 Expression level STK33 gene in cancer tissues and distal cancerous tissues of patients with lung cancer(±s)
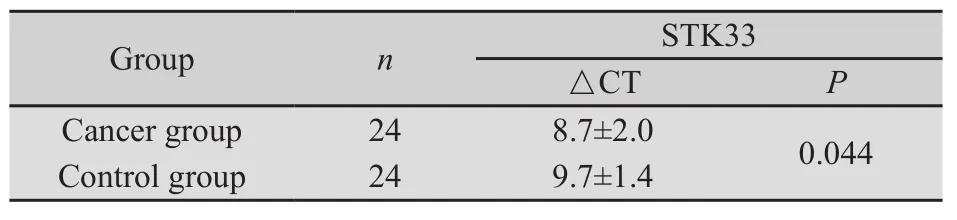
表1 肺癌患者肺癌组织和远癌组织中STK33水平Tab.1 Expression level STK33 gene in cancer tissues and distal cancerous tissues of patients with lung cancer(±s)
2.1.3.2 NSCLC患者肺癌组织与肺良性病变患者组织中STK33 mRNA的表达情况 实时荧光定量PCR结果显示,NSCLC患者肺癌组织与良性病变患者组织中STK33平均ΔCT值分别为8.7±2.0和10.5±0.9。NSCLC患者肺癌组织STK33基因表达量高于肺良性病变组织,且两者比较差异有统计学意义(P<0.05,表 2)。
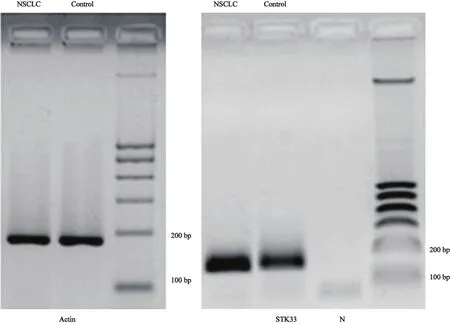
图2 NSCLC和对照组组织中β-actin、STK33 real-time RT-PCR产物琼脂糖凝胶电泳图Fig.2 Agarose gel eletrophoretogram of real-time RT-PCR products of actin, STK33 in NSCLC and control group tissues
表2 肺癌患者肺癌组织与良性病变患者组织中STK33水平Tab.2 Expression level STK33 gene in tissues of patients with lung cancer and lung benign disease(±s)

表2 肺癌患者肺癌组织与良性病变患者组织中STK33水平Tab.2 Expression level STK33 gene in tissues of patients with lung cancer and lung benign disease(±s)
Group n STK33△CT P Distal cancerous group 24 8.7±2.0 Lung benign disease group 9 10.5±0.9 0.001
2.1.3.3 NSCLC患者组织中STK33 mRNA表达与临床病理因素的关系 t检验及单因素方差分析显示,STK33 mRNA的表达量与NSCLC患者的性别、年龄、吸烟史、肿瘤大小、组织学类型、分化程度、分期、淋巴结转移无关(P>0.05,表3)。
2.2 蛋白印迹法检测STK33蛋白表达情况的结果分析 灰度分析结果显示STK33在NSCLC患者的肺癌组织中的蛋白表达,明显高于远癌组和肺良性病变组(图3、4)。
3 讨 论
肿瘤发生发展的过程中往往有着多重复杂的遗传和后天的异常改变,尽管存在着这种复杂性,有些肿瘤的形成和存活总是倾向于被某一特定的癌基因所驱动[12]。因此当我们灭活或钝化这一特定癌基因,就可以影响到肿瘤细胞的存活、分化、阻滞或者衰老。这体现了肿瘤靶向治疗方法的核心概念就是在肿瘤形成的癌基因网络中,寻找到关键点加以抑制,使其整个系统崩溃,并最大程度地保护正常细胞[13]。
但是实际情况,很多针对癌基因的靶向治疗,其临床疗效并不乐观。原因在于肿瘤的发生、演变过程是多因素、多通路、多阶段的复杂过程。尽管如此,其中有一个误区仍需要澄清,并不是所有与肿瘤发生、发展相关的基因和通路都是固有致癌的[14]。值得注意的一点就是,相对于正常组织细胞来说,这些非致癌的基因和通路对于维持肿瘤发生、发展是不可或缺的。进一步来说,当肿瘤发生、发展所需要的癌基因异常时,肿瘤细胞会启动后备的非癌基因,以维持其活性。随即肿瘤细胞对非癌基因产生依赖性的现象,称之为非癌基因成癌(non-oncogene addiction,NOA)。这样一来,干扰这些基因可能为肿瘤靶向治疗提供了新的机会[15]。

表3 肺癌患者STK33 mRNA的表达量与肺癌的临床病理特征的关系Tab.3 Relationship between expression level STK33 gene and clinical pathology characteristics in the lung cancer tissues
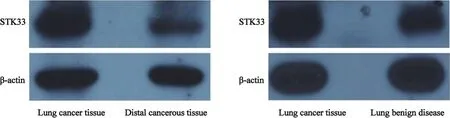
图3 Western blot 比较肺癌组织、远癌组织和肺良性病变组织中STK33和β-actin蛋白表达情况Fig.3 Expression of STK33 and β-actin protein in NSCLC cases group, distal cancerous tissues group and lung benign disease cases group detected by Western blot

图4 灰度分析所得NSCLC、远癌组组织和肺良性病变组织中STK33灰度值与相应组织中的β-actin的灰度值比较Fig.4 Expression of STK33 protein and β-actin in NSCLC cases group, distal cancerous tissues group and lung benign disease cases group using gray intensity analysis
在这样的理论背景下,Scholl等[6]研究发现与突变的K-ras基因依赖的相关肿瘤细胞系中,通过抑制STK33能产生综合致死效应,而对于那些非K-ras依赖的细胞系却没有作用。同时通过检测肿瘤细胞系、人体骨髓和末梢血标本的研究进一步指出,STK33没有出现类似癌基因表现般的结构异常,且在K-ras突变和不突变不同背景下的表达水平没有显著性差异。提示STK33是作为非癌基因发挥功效的,这点和上文提到的非癌基因成瘾的理念是一致的。而且最近一些研究表明某些非癌基因对于某些特定的肿瘤类型有特异性,如多发性骨髓瘤、乳腺癌和结肠癌[16-18]。这可能反映了不同细胞谱系对生长和生存的不同需要。提示在NSCLC中尤其存在K-ras突变的背景下,非癌基因STK33对于肺癌的发生、发展会不会也有特异性呢?即使不是特异性,NSCLC的发生、发展对于非癌基因STK33的依赖性也应值得注意。Scholl等[6]的研究就巧妙地解决了TKI耐药性的困境。我们可以想象STK33抑制剂作为一种治疗策略来干预K-ras突变的NSCLC患者的美好前景。而我们研究工作的意义就在于初步解决了TKI耐药性的基础问题,即NSCLC的发生、发展需要STK33的参与。
本研究着手探讨STK33在非小细胞肺癌发生、发展中的临床意义颇具针对性。本项目中我们采用实时荧光定量PCR法来检测STK33 mRNA在NSCLC中的表达。研究观察到NSCLC患者肺癌组织中STK33基因mRNA平均相对表达量高于远癌组织和肺良性病变中STK33基因的表达水平。那么STK33基因在肺癌组织与对照组之间的这种差异性表达提示STK33在NSCLC的发生、发展中的影响不容忽视。接着,我们进一步在NSCLC患者肺癌组织和对照组织中的STK33基因表达量存有差异的样本中,应用Western blot法来验证STK33蛋白表达情况,结果发现肺癌组织、远癌组织和肺良性病变组织中STK33蛋白表达也存在差异。这些实验结果暗示着STK33基因与肺癌发生、发展之间有密切关联。同时结合临床病理数据分析显示,STK33 mRNA的表达量与NSCLC患者的性别、年龄、吸烟史、肿瘤大小、组织学类型、分化程度、分期和淋巴结转移都无关(P>0.05),提示STK33能否作为肺癌的重要分子标志物还需进一步研究,因此我们将继续深入探索相关的未知领域。值得注意的是,以60岁分界的高年龄组与低年龄组之间有差异的趋势,但这种差异没有统计学意义,具体原因有待进一步研究。
综上所述,本研究初步揭示了STK33与NSCLC之间潜在的密切关联。为干扰STK33从而影响肿瘤发生、发展的研究路线提供了理论支持。为肿瘤靶向治疗位点的巧妙选择,非癌基因成瘾现象的灵活实践提供了新思路。
[1] Jemal A, Siegel R, Ward E, et al.Cancer statistics, 2006[J].CA Cancer J Clin, 2006, 56: 106-130.
[2] Giaccone G.Epidermal growth factor receptor inhibitors in the treatment of non-small cell lung cancer[J].J Clin Oncol,2005, 23: 3235-3242.
[3] Pao W, Wang TY, Riely GJ.K-ras mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib[J].PLoS Med, 2005, 2:e17.
[4] Massarelli E, Varella-Garcia M, Tang X, et al.K-ras mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer[J].Clin Cancer Res, 2007, 13:2890-2896.
[5] Jackman DM, Sequist LV, Cioffredi L, et al.Impact of EGFR and K-ras genotype on outcomes in a clinical trial registry of NSCLC patients initially treated with erlotinib or gefitinib[C].J Clin Oncol, 2008: 26 (abstr 8035).
[6] Scholl C, Frohling S, Dunn IF, et al.High-throughput RNA interference screening identifies synthetic lethality between oncogenic K-ras dependency and suppression of STK33[C].Blood:ASH Annual Meeting Abstracts, 2008, 112: 3806.
[7] Mujica A, Hankeln T, Schmidt ER.A novel serine/threonine kinase gene, STK33, on human chromosome 11p15.3 [J].Gene, 2001, 280:175-181.
[8] Amid C, Bahr A, Mujica A, et al.Comparative genomic sequencing reveals a strikingly similar architecture of a conserved syntenic region on human chromosome 11p15.3(including gene ST5) and mouse chromosome 7[J].Cytogenet Cell Genet, 2001, 93: 284-290.
[9] Manning G, Whyte DB, Martinez R, et al.The protein kinase complement of the human genome [J].Science, 2002, 298:1912-1934.
[10] Kostich M, English J, Madison V, et al.Human members of the eukaryotic protein kinase Family [J].Genome Biol, 2002,3(9): 43.
[11] Caenepeel S, Charydczak G, Sudarsanam S, et al.The mouse kinome: Discovery and comparative genomics of all mouse protein kinases [J].Proc Natl Acad Sci USA, 2004, 11707-11712.
[12] Weinstein IB.Cancer.Addiction to oncogenes-the achilles heal of cancer [J].Science, 2002, 297:63-64.
[13] Luo J, Solimini NL, Elledge SJ.Principles of cancer therapy:oncogene and non-oncogene addiction [J].Cell, 2009, 136:823-837.
[14] Solimini NL, Luo J, Elledge SJ.Non-oncogene addiction and the stress phenotype of cancer cells [J].Cell, 2007, 130:986-988.
[15] Kaelin WG Jr.The concept of synthetic lethality in the context of anticancer therapy [J].Nat Rev Cancer, 2005, 5: 689-698.
[16] Schlabach MR, Luo J, Solimini NL, et al.Cancer proliferation gene discovery through functional genomics [J].Science,2008, 319: 620-624.
[17] Shaffer AL, Emre NC, Lamy L, et al.IRF4 addiction in multiple myeloma [J].Nature, 2008, 454: 226-231.
[18] Silva JM, Marran K, Parker JS, et al.Profiling essential genes in human mammary cells by multiplex RNAi screening [J].Science, 2008, 319:617-620.
