淫羊藿黄酮的高效液相色谱分析方法
2010-09-19朱靖博,邱焕杰,李丹凤
朱 靖 博,邱 焕 杰,李 丹 凤
(大连工业大学 生物与食品工程学院,辽宁 大连 116034)
0 引言
淫羊藿(EpimediumHerb)又名仙灵脾,始载于《神农本草经》。本品辛、甘,温,入肝、肾经,是我国传统补益类中药[1]。临床上,淫羊藿黄酮类化合物用于治疗骨质疏松症的效果尤其显著[2-4]。淫羊藿属植物中的活性成分以黄酮类成分为主,大部分为8位具有异戊烯基取代的淫羊藿苷类黄酮成分[5]。2005 年版《中国药典》规定淫羊藿药材质量的控制指标为淫羊藿苷。
高效液相色谱技术被认为是中药质量控制的有效手段,黄朝瑜等以0.5%冰醋酸溶液和乙腈为流动相,建立了淫羊藿药材的指纹图谱,共检出7个峰[6]。郭青等得到了包括朝霍定A、B、C 以及淫羊藿苷一组峰的指纹图谱[7]。易中宏等以乙腈-水-36%醋酸溶液梯度洗脱的HPLC法比较研究巫山淫羊藿的不同部位[8]。但由于微量成分分离和标准样品缺乏等方面原因,以往HPLC 分析中对每个黄酮的定量分析研究较少。王丽霞以淫羊藿苷为内标,对不同淫羊藿属9个共有峰做了相对峰面积的分析[9],但未进行定量分析。
利用HPLC对样品每个色谱峰进行定性、定量分析已成为中药质量控制的主要手段[12]。但标准对照品的缺乏是制约这类方法应用于定量分析的主要因素。本实验旨在建立以淫羊藿苷标准品为参照物,对其他淫羊藿黄酮进行相对定量,得到其他淫羊藿黄酮的相对含量,从而得到淫羊藿总黄酮的相对含量的方法,以期在一定程度上反映淫羊藿水提物的内在质量,为控制以淫羊藿黄酮为活性物质的药材及药品质量提供参考。
1 实验
1.1 仪器与试剂
DIONEX UltiMate3000高效液相色谱仪,美国戴安公司;P680 泵;ASI1000自动进样器;PDA100检测器;1810D 型自动双重纯水蒸馏器,上海申生科技有限公司;UV-2102PC紫外可见光分光光度计,尤尼柯上海仪器有限公司。
乙腈,色谱纯;甲醇、乙醇,分析纯;水,双重蒸馏水。
1.2 药 品
淫羊藿苷对照品,由中国药品生物制品检定所提供;淫羊藿提取物,购自西安保赛生物工程有限公司。
1.3 色谱条件
色谱柱:Sinochrom ODS-BP(4.6mm×250mm,5μm);流动相:A 为乙腈,B 为双重蒸馏水;乙腈浓度梯度:0~12min(20%~30%),12~20min(30%~36%),20~30 min(36%~58%);洗脱时间:30min;体积流量:1.0mL/min;检测波长:270nm;进样量:20μL。
1.4 对照品溶液的制备
精确称取淫羊藿苷标准品1.12mg于10mL容量瓶中,甲醇定容,质量浓度为112μg/mL。分别取一定量标准品溶液稀释不同倍数,配成质量浓度为70、56、22.4、11.2、2.24μg/mL 的溶液,按“1.3”色谱条件分别进样分析。
1.5 供试品溶液的制备
称取淫羊藿水提物50g放置于2 000mL圆底烧瓶中,加入80%乙醇500mL,60℃水浴锅中提取2次,每次2h,过滤,合并滤液浓缩,10%乙醇溶解定容于500 mL 容量瓶,得10 mg/mL 溶液,用该溶液配制1mg/mL 溶液,0.45μm 微孔滤膜过滤,作为供试品溶液备用。
1.6 检测波长的选择
对照品溶液在紫外分光光度仪上进行扫描(190~400nm),确定淫羊藿黄酮类化合物的最大紫外吸收波长。
2 结果与分析
2.1 淫羊藿苷的紫外吸收光谱与标准曲线
对照品溶液紫外吸收光谱扫描的结果见图1(190~400nm),从而确定淫羊藿黄酮类化合物的紫外吸收光谱最大吸收在为270nm。
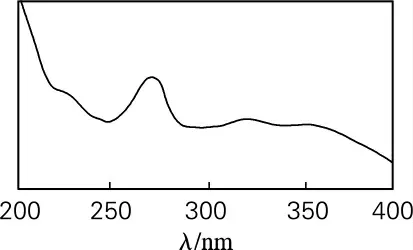
图1 淫羊藿苷紫外吸收光谱图Fig.1 Ultraviolet absorption spectrum of Icariin
对“1.4”制得的对照品溶液,通过液相色谱检测得到标准曲线线形回归方程:y=0.633 2x-0.390 8,R2=0.999 9。结果表明,在2.24~112μg/mL淫羊藿苷峰面积与质量浓度呈良好线性关系。
2.2 样品分析
淫羊藿苷和供试品溶液色谱分离结果如图2、3所示。图2中淫羊藿苷纯度为98.5%。该色谱条件对淫羊藿水提物进行分离得到13个分离良好的色谱峰。图3中的9号峰为淫羊藿苷。
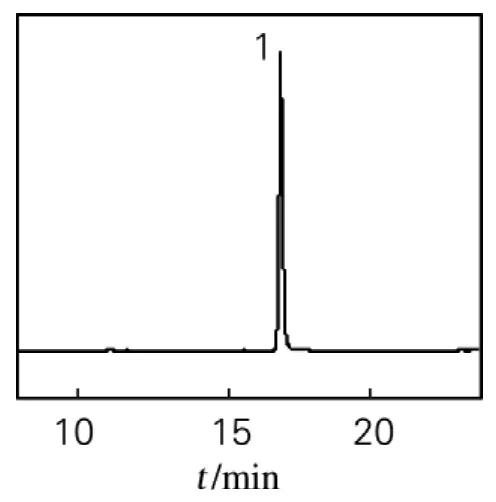
图2 淫羊藿苷HPLC图Fig.2 Chromatogram of Icariin

图3 淫羊藿黄酮HPLC图Fig.3 Chromatogram of Epimedium Flavones
每种物质都有其特定的紫外吸收光谱,其紫外吸收光谱由物质的结构决定,类似结构的物质其紫外吸收光谱具有相似性,所以紫外吸收光谱可作为定性的依据之一[10-11]。利用HPLC-PDA检测13个色谱峰的紫外吸收光谱图见图4。从图1和图4可以看出,在该色谱条件下分离的13个峰,其紫外吸收光谱吸收与淫羊藿苷紫外吸收相同,可以推测这13个化合物都是淫羊藿黄酮类化合物。

图4 淫羊藿黄酮HPLC1~13色谱峰紫外吸收波谱图Fig.4 Ultraviolet absorption spectrum of peaks 1-13 of Epimedium Flavones Chromatogram
2.3 分析方法学考察
2.3.1 稳定性试验
将供试品溶液在室温下放置,按“1.3”色谱条件,分别在制备供试品后的0、5、10、15、20h,吸取20μL注入高效液相色谱仪,记录淫羊藿13个黄酮化合物的峰面积和保留时间,计算相对标准偏差。峰面积RSD为1.43%~4.03%,保留时间RSD为0.011%~0.65%。结果表明样品20h内是稳定的。
2.3.2 精密度试验
供试品溶液按“1.3”色谱条件连续进样5次,记录淫羊藿13个黄酮化合物的峰面积,计算相对标准偏差。峰面积RSD为1.28%~4.16%,结果表明该方法具有良好的精密度。
2.3.3 重现性试验
按照“1.5”供试品溶液制备方法,重复制备5份样品,按“1.3”色谱条件测定,记录淫羊藿13个黄酮化合物的峰面积和保留时间,计算相对标准偏差。峰面积RSD为0.35%~3.67%,保留时间RSD为0.014%~0.46%。结果表明该方法的重现性良好。
2.3.4 回收率试验
移液管吸取1 mg/mL 的样品1、1、2、2 mL于5、10、25、50 mL 容量瓶,用甲醇定容,即样品分别稀释了5、10、12.5、25 倍,再用移液管各取1mL分别与1 mL 的26μg/mL 的淫羊藿苷混溶,按“1.3”色谱条件测定,记录淫羊藿苷的峰面积和样品分析回收率,结果如表1所示。

表1 淫羊藿苷回收率测定结果Tab.1 The recovery rate of Icariin
2.3.5 各黄酮单体定量分析
取供试品溶液按“1.3”色谱条件连续进样4次,记录淫羊藿13个色谱峰的峰面积,代入线性回归方程,即可得到每个黄酮单体相当于淫羊藿甙的含量。结果如表2所示。
试验表明该淫羊藿提取物总黄酮质量浓度约为244.184μg/mL,淫羊藿溶液的质量浓度为1mg/mL,即淫羊藿提取物中以淫羊藿苷计的总黄酮质量分数为24.4%。
3 结论
(1)通过试验研究,建立了淫羊藿黄酮化合物HPLC分析方法。该方法分析时间短,样品分离度高,精密度、稳定性和重现性好,为淫羊藿水提物及其制剂的质量控制提供了依据。
(2)以HPLC-PDA 联用方法,确定这13 个峰都是淫羊藿黄酮化合物色谱峰。
(3)以淫羊藿苷为标准品,对这13个黄酮化合物进行了相对定量分析,结果显示提取物以淫羊藿苷计的总黄酮质量分数为样品总量的24.4%,该指标可作为控制淫羊藿水提物质量的参考。

表2 淫羊藿提取物中黄酮的质量浓度Tab.2 The content of Icariin in Epimedium extract μg·mL-1
