丙酮-肼基二硫代甲酸苄酯的合成、晶体结构及量子化学研究
2010-09-14季宁宁
季宁宁
(泰山学院化学与环境科学系,山东泰安 271021)
丙酮-肼基二硫代甲酸苄酯的合成、晶体结构及量子化学研究
季宁宁
(泰山学院化学与环境科学系,山东泰安 271021)
用密度泛函理论(DFT)B3LYP方法选取6-31G*基组,运用Gaussian03量子化学程序包,对丙酮-肼基二硫代甲酸苄酯晶体进行了量子化学计算研究,探讨了化合物的稳定性、各原子净电荷分布、一些前沿的分子轨道能量和组成特征.计算结果与通过单晶X-射线衍射法测得的晶体结构的实验结果吻合.此项研究可为该配体及其金属配合物的合成及实际应用提供理论参考.
密度泛函理论(DFT);丙酮-肼基二硫代甲酸苄酯晶体;量子化学计算
0 引言
近年来,由于含硫席夫碱及其金属配合物具有良好的抑菌、抗疟疾、抗肿瘤和抗病毒等生物活性而引起了化学和药学界的广泛关注[1-6].近年来在这方面的研究工作十分活跃,如A li[7]课题组制备得到了丙酮-肼基二硫代甲酸苄酯晶体,并以该晶体为配体合成了锡的过渡金属配合物;我们课题组[8]也以该配体为起始物合成了金属铜的配合物,虽然该类配体已经合成并被报道,但目前尚没有该类分子电子结构与配位性能关系的研究报道.为了探讨该类化合物的电子结构特征与配位性能的关系,更有效地指导新的过渡金属配合物的设计工作,基于此,作者对丙酮-肼基二硫代甲酸苄酯晶体进行了B 3LYP/6-31G*水平上的密度泛函量子化学计算,获取了分子的几何构型、原子的净电荷密度、前沿分子轨道特征组成、能级与轨道电子密度等参数,初步探讨了这些参数与配位性能间的关系.
1 实验部分
1.1 仪器与试剂
元素分析用PE-2400 II型元素分析仪测定,红外光谱用N ico let6700型红外光谱仪(KB r压片)测定,熔点测定用X 4型显微熔点仪(温度计未校正)测定,晶体结构在B ruker Sm art APEX IICCD型单晶测试系统中测定,所用试剂均为AR级.
1.2 标题化合物单晶制备
按照文献[9]的方法制备了标题化合物,并培养得到了晶体.m.p.110~111℃.IR(KB r)ν:3165, 1624,1481,1312,1245,1066,999,723cm-1.A nal.calcd fo r C11H14N2S2:C55.42,H 5.92,N 11.75;found C55.26,H6.12,N11.97.
1.3 晶体结构测定
选取尺寸为0.15mm×0.12mm×0.10mm的晶体置于B ruker Sm artAPEX IICCD型X射线单晶衍射仪上,以石墨单色化的Mo Kα(λ=0.071073nm)辐射为光源,在室温(295±2)K下,以面探扫描方式扫描,在1.71°≤θ≤25.05°范围内收集6266个强衍射点,其中独立衍射点2200个(Rint=0.0216),I> 2σ(I)的可观测衍射点为1865个.晶体结构由直接法解出,非氢原子的坐标是在以后的数轮差值Fourier合成中陆续确定的,对全部非氢原子的坐标及各向异性参数用SHELXL-97程序以最小二乘法对F2进行精修,其中的非氢原子都作了各向异性修正,碳上的氢原子根据理论加氢获得.最终偏离因子R1=0.0322,wR2=0.0841;w=1/[σ2(Fo2)+(0.0450P)2+0.3042P],其中P=(Fo2+2Fc2)/3;(Δρ)max=0.293e.Å-3,(Δρ)min=-0.337e.Å-3.标题化合物的晶体学数据列于表1.

表1 标题化合物的晶体结构数据Tab le 1 C rysta l da ta and struc tu re ref inem en ts of the title com pound
1.4 DFT计算
根据标题化合物晶体结构的各原子坐标位置,取一个标题化合物结构单元,在B3LYP/6-31G*基组水平上,进行了量子化学单点计算,计算采用的原子输入坐标来自该晶体结构数据,计算涉及29个原子,261个原子基本函数,524个初始高斯函数,其中有63个占据轨道,63个α电子及63个β电子,计算体系的电荷为0,多重度为1.所有计算均运用Gaussian 03W程序包完成.
2 结果与讨论
2.1 标题化合物的晶体结构
图1为标题化合物的分子结构图,图2为二聚体结构图,图3为晶胞堆积图,氢键列于表2.
由图2和表3可知,标题化合物相邻分子间H1-S1键长为2.72Å,这个键长比两原子间的共价键(1.37Å)要长的多,但比范德华半径之和(3.00Å)短[10],因此可看作是弱的氢键作用,通过该弱的N–H…S氢键形成了一个二聚体结构.

表2 标题化合物的氢键键长(Å)和键角(°)Tab le 2 Hydrogen bond lengths(Å)and ang les(°)for the title com pound

图1 标题化合物的分子结构Figure 1 M o lecu lar structure of the title com pound


表3 标题化合物的量子化学计算值与X-射线单晶衍射实验值的主要键长(Å)和键角(°)比较Tab le 3 The com pa r ison of bond lengths(Å)and ang les(°)of ca lcu la ted and exper im en t va lue of quan tum chem istry
2.2 标题化合物的几何构型
表3是量子化学计算所得的键长和键角数据与X-射线单晶衍射的实验值的比较,图4是标题化合物的优化构型.由图4对比标题化合物的分子结构图1可以看出二者构型基本吻合;由表3可以看出,计算得到的键长绝大多数都比实验值稍长,这是因为计算的键长属于气态孤立分子,而实验键长对应于晶体分子,计算得到的键长和实验值相差最大为0.1574Å(N(1)-H(1)).计算得到的键角和实验值相差最大为4.2194°(C(4)-N(1)-H(1)),计算得到的键长键角数据和从晶体结构得到的数据符合的很好,表明用B3LYP/6-31G*计算丙酮-肼基二硫代甲酸苄酯晶体的分子几何构型是可靠的,所有计算结果令人满意.

图4 标题化合物的优化分子模型(B3LYP/6-31G*)Figure 4 O p tim ized structure of the title com pound by B3LYP/6-31G*
2.3 标题化合物的能量和分子轨道组成
计算得到标题化合物体系总能量为-1333.46389a.u,占据轨道的能量HOMO为-0.21373a.u.,非占轨道LUMO的能量为-0.04648a.u..由于体系能量与占据前沿轨道的能量均较低,表明该标题化合物基态较为稳定,这与实验结果是一致的.从氧化还原或电荷转移分析,由于两前沿轨道间的能量间隙仅为1.09082a.u.,该标题化合物基态容易失去电子而被氧化,因此氧化还原稳定性较差.
为探索该标题化合物的电子结构与成键特征,对标题化合物分子轨道进行了系统分析,用参与组合的各类原子轨道系数的平方和表示其在分子轨道中的贡献,并经归一化.把标题化合物原子分为六部分:(a)S原子;(b)N原子;(c)苯环C(Ⅰ)原子;(d)双键C(Ⅱ)原子;(e)其它C(Ⅲ)原子;(f)氢原子H.分别讨论了5个前沿占据轨道和5个前沿非占轨道,计算结果如表4和图4所示.

表4 标题化合物的分子轨道组成(%)Tab le4 Som e ca lcu la ted fron tierm olecu la r orb ita l com position(%)of title com pound
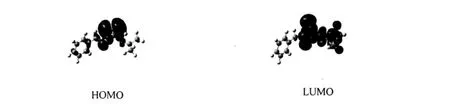
图5 标题化合物的前沿分子轨道示意图Figure 5 Schem a tic d iagram of the fron tierMO for the title com pound
由表4和图5可知,标题化合物的HOMO主要由S原子和N原子组成,其中S原子达82.39%,N原子10.47%,其它原子对HOMO的贡献均较少,C(Ⅰ)原子为2.81%,C(Ⅱ)原子为1.19%,C(Ⅲ)原子为2.35%,H原子为0.69%,表明S原子和N原子比较容易发挥供电子作用,在形成金属配合物时可提供电子形成配位键;而在最低未占轨道,各原子对LUMO的贡献变化比较明显,LUMO主要由S原子、N原子、C(Ⅱ)原子和H原子组成,其中C(Ⅱ)原子贡献最大,达50.11%,S原子为25.54%,N原子10.68%,H原子11.82%,其它原子则贡献较小,C(Ⅰ)原子为0.75%,C(Ⅲ)原子为1.10%.比较HOMO与LUMO的轨道成分,发现标题化合物从基态向激发态电子转移时,主要是S原子上的电子向C (Ⅱ)原子和H原子(主要是甲基C上的H原子)轨道转移,形成电荷转移激发态配合物.
2.4 标题化合物的电子结构布局

表5 标题化合物的原子电荷分析(B3LYP/6-31G*)Tab le5 The a tom ic charge popu la tion by B3LYP/6-31G*level
表5所示为标题化合物各原子净电荷的M u lliken布局分析.结果表明:所有氢原子、不连氢的碳原子和单键硫原子均带正电荷;与氢相连的碳原子、氮原子和双键硫原子均带负电荷.在分子中,非氢原子中荷正电较大的是S(2)、C(2)和C(6),荷正电荷值分别为0.224834、0.330311和0.142815;荷负电较大的原子有S(1)、N(1)、N(2)、C(1)、C(3)和C(5),负电荷值分别为-0.255282、-0.398539、-0.553179、-0.510184和-0.515465,这主要是由原子的电负性决定的,如果原子周围连有电负性较大的氮和硫等原子,则该原子荷正电,而氮和硫原子荷负电;与氢相连的原子它们较强烈地吸引键合氢原子上的电荷,使其自身带上负电荷而相连的氢原子带上正电荷,由于S(1)和N(1)荷负电较多,因此它们是标题化合物的化学活性部位,它们在形成配合物时可作为配位原子,这与文献报道是一致的.
3 结论
利用量子化学的密度泛函理论(DFT)B3LYP方法,对标题化合物的晶体结构进行优化,计算了单点能、分子轨道组成和原子净电荷的M u lliken布局,较好地阐明了标题化合物的稳定性与成键特征,与实验结果取得了很好的一致,为以后以该化合物为配体的金属配合物的设计提供了理论参考.
[1]鲁传华,周双生.含硫Schiff碱铜(Ⅱ)配合物的合成与表征[J].化学试剂,2002,24(4):227-228.
[2]杜家声,李自弘,黄渊泽,等.含硫希夫碱配合物的研究Ⅱ[J].无机化学学报,1994,10(1):47-52.
[3]AkbarA M,Teoh SG..M agnetic and Spectroscop ic Studies on M etal Comp lexesof som eOxygen-N itrogen and Sulphur-N itrogen Chelating Agents[J].Journalof Inorganic and NuclearChem istry,1978,40(3):451-458.
[4]IskanderM F,EI-Sayed L,EI-Toukhy,etal.Reac tivity of some coordinated ligands containing sulphur towards nucleophilic substitution reactions.Part I.Reaction of[(substituted 1,2-ethanediy lidene)bis(S-m ethy lhydrazine-carbodithioate)-N,N′,S,S′,(-2)]nickel (II)and palladium(II)chelatesw ith secondary am ines[J].Inorganica Chim ica A cta,1986,123(1):19-25.
[5]Subhash P,George B K..TransitionM etalComp lexesof Sem icarbazonesand Thiosem icarbazones[J].Coordination Chem istry Review s, 1985,63:127-160.
[6]郑启升,赵吉寿,王金城,等.含硫双Schiff碱及其金属配合物的合成与生物活性研究[J].云南民族大学学报(自然科学版), 2007,16(3):243-246.
[7]A liM A,BakerH JH A,M irzaA.H.,etal.Preparation,spec troscopic characterization and X-ray crystalandmolecular structuresof nickel(II),copper(II)and zinc(II)comp lexesof the Schiff base form ed from isatin and S-m ethyldithiocarbazate(H isa-sm e)[J].Polyhedron,2008,27(1):71-79.
[8]季宁宁,石智强,郑泽宝,等.含S,N的配体及其铜配合物的合成与晶体结构[J].合成化学,2009,17(4):432-436.
[9]A liM A,M irzaA H,Bu tcherR J,etal.Biologicalactivity ofpalladium(II)and p latinum(II)comp lexesof the acetone Schiffbasesof S-methyl-and S-benzyldithiocarbazate and the X-ray crystal struc ture of the[Pd(asm e)2](asm e=anionic form of the acetone Schiff base of S-m ethyldithiocarbazate)comp lex[J].J Inorg B iochem,2002,92:141-148.
[10]BondiA.Van derW aalsVolum es and Radii[J].JPhysChem,1964,68(3):441-451.
Syn thesis,C rysta l Structure and Quan tum Chem istry of N’-Isopropylidene
-hydrazinecarbod ith ioic ac id benzyl
JIN ing-ning
(Departm en tof Chem istry and Environm ental Science,Taishan University,Tai’an,271021,China)
The N’-Isop ropylidene-hyd razinecarbodithioic ac id benzyl esterwas sub jected to density functional theory(DFT)calcu lations using B3LYP/6-31G*basis set.The stabilitiesof the compound,the orbital energies,M u lliken charge distribution and composition characteristicsof som e frontiermo lecu laro rbitalshave been investigated.The theo retical resu ltsacco rdedw ith experim ental resu ltsof the crystal st ructure of the title compound by X-ray d iffraction analysis.The research served asa theoretical reference for the p reparation and p ractical app lication of the ligand and its comp lexes
density functional theo ry(DFT);N’-Isop ropylidene-hyd razinecarbodithioic acid benzyl ester;quantum chem istry calcu lation
O64
A
1672-2590(2010)03-0090-06
2010-04-15
泰山学院人才引进项目(Y06-2-08)
季宁宁(1979-),女,山东烟台人,泰山学院化学与环境科学系讲师.
