HPLC法测定不同来源甘草中甘草酸的含量Δ
2010-09-10唐丽琴张善堂石允卉安徽医科大学附属省立医院药剂科合肥市230001
唐丽琴,张善堂,刘 圣,石允卉(安徽医科大学附属省立医院药剂科,合肥市 230001)
HPLC法测定不同来源甘草中甘草酸的含量Δ
唐丽琴*,张善堂,刘 圣,石允卉(安徽医科大学附属省立医院药剂科,合肥市 230001)
目的:建立以高效液相色谱法测定甘草中有效成分甘草酸含量的方法,并测定不同产地和基原甘草药材中甘草酸的含量。方法:色谱柱为Hypersil-C18(250 mm×4.6 mm,5µm),流动相为甲醇-0.2 mol·L-1醋酸铵溶液-冰醋酸-三乙胺(64∶36∶1.5∶0.02),流速为1.0 mL·min-1,检测波长为252 nm,柱温为30℃。结果:甘草酸检测浓度在0.041 6~0.208 0 mg·mL-1范围内与峰面积积分值呈良好线性关系(r=0.999 9);平均回收率为101.3%,RSD=1.22%(n=9)。结论:不同来源甘草药材中甘草酸的含量差异较大,野生甘草中甘草酸的含量高于家种。
甘草;甘草酸;高效液相色谱法;含量;来源
Δ安徽省自然科学基金面上项目(090413106)
*副主任药师,博士。研究方向:临床药学、药理学。电话:0551-2283378-809。E-mail:tulcyl@vip.sina.com
甘草为豆科植物甘草Glycyrrhiza uralensisFisch.、胀果甘草G.inflateBat.或光果甘草G.gliabraL.的干燥根及根茎,具补脾益气、清热解毒、祛痰止咳、缓急止痛、调和诸药之功效[1]。甘草酸是其最重要的活性成分之一[2~4],具有抗炎、抗溃疡、抗肝毒素以及抗癌等药理作用[5,6]。但商品甘草的产地、基原等来源复杂,其有效成分甘草酸的含量也存在很大差异,这给医院临床用药造成一定影响。目前,国内对不同来源甘草饮片有效成分的系统研究较少,为此本研究以甘草酸为检测指标,采用高效液相色谱(HPLC)法对不同产地不同基原的甘草进行初步分析研究,以期为医院的甘草饮片采购提供选择依据,从而为临床提供更佳的药材。
1 仪器与材料
1.1 仪器
LC-6A型HPLC仪,包括SPD-6AV紫外-可见检测器、HS色谱数据工作站(日本岛津公司);ZG-1型打粉机(上海淀元实业有限公司);BT124S型电子分析天平(北京赛多利斯仪器系统有限公司);JK-DY500型医用数控超声清洗器(合肥金尼克机械制造有限公司)。
1.2 试药
甘草酸单铵盐对照品(中国药品生物制品检定所,批号:110731-200614);甲醇为色谱纯,冰醋酸、醋酸铵、三乙胺为分析纯,水为重蒸馏水;甘草药材均为从当地购买的市售药材,并经安徽中医学院药学院刘守金教授鉴定为真品。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Hypersil-C18(250 mm×4.6 mm,5µm);流动相:甲醇-0.2 mol·L-1醋酸铵溶液-冰醋酸-三乙胺(64∶36∶1.5∶0.02);检测波长:252 nm;柱温:30 ℃;流速:1.0 mL·min-1;进样量:20 μL。理论板数按甘草酸峰计算应不低于2 000。色谱见图1。
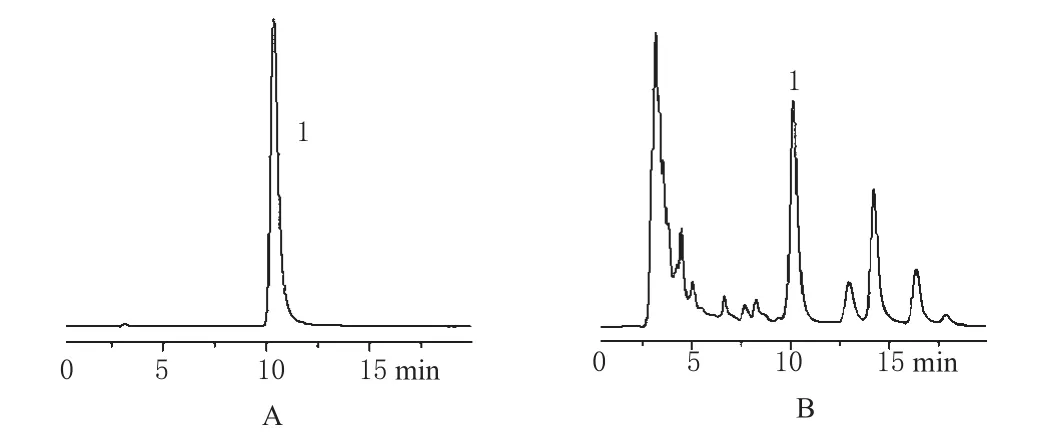
图1 高效液相色谱图A.对照品;B.甘草药材;1.甘草酸单铵盐Fig 1 HPLC chromatogramsA.reference substance;B.G.uralensis;1.monoammonium glycyrrhizinate
2.2 对照品溶液的制备
称取甘草酸单铵盐对照品约10 mg,精密称定,置于10 mL容量瓶中,加流动相稀释至刻度,摇匀,制成浓度为1.06 mg·mL-1的甘草酸单铵盐溶液,折合甘草酸为1.04 mg·mL-1,作为对照品贮备液。再精密量取对照品贮备液0.4、0.8、1.2、1.6、2.0 mL,置于10 mL容量瓶中,加流动相稀释至刻度,摇匀,得到含甘草酸分别为 0.041 6、0.083 2、0.124 8、0.166 4、0.208 0 mg·mL-1的对照品溶液。
2.3 供试品溶液的制备
取甘草药材粉末约0.2 g,精密称定,置于50 mL容量瓶中,加入流动相适量,超声提取45 min,静置,用流动相定容,摇匀,用0.45 μm微孔滤膜滤过,即得。
2.4 线性关系考察
分别精密吸取“2.2”项下各对照品溶液20 μL,按上述色谱条件进行测定。以峰面积积分值(A)对对照品浓度(C)进行线性回归,得回归方程为A=9.0×106C+2 072.7(r=0.999 9)。结果表明,甘草酸检测浓度在0.041 6~0.208 0 mg·mL-1范围内与峰面积积分值呈良好的线性关系。
2.5 精密度试验
精密吸取浓度为0.124 8 mg·mL-1的对照品溶液20 μL,按上述色谱条件连续进样6次,测定峰面积。结果,RSD=0.52%,表明仪器精密度良好。
2.6 稳定性试验
精密量取同一供试品溶液20 μL,分别在0、1、2、3、4、5、24 h进样测定。结果,RSD=1.07%,表明供试品溶液在24 h内稳定。
2.7 重复性试验
称取同一甘草样品粗粉约0.2 g,平行6份,精密称定,按“2.3”项下方法制备供试品溶液。精密吸取20 μL,按上述色谱条件测定并计算甘草酸含量。结果,RSD=1.11%,表明方法重复性较好。
2.8 加样回收率试验
取已知含量的甘草样品9份,每份约0.1 g,精密称定,加入相应量的对照品,按“2.3”项下方法制备供试品溶液,按上述色谱条件测定,计算加样回收率,结果见表1。
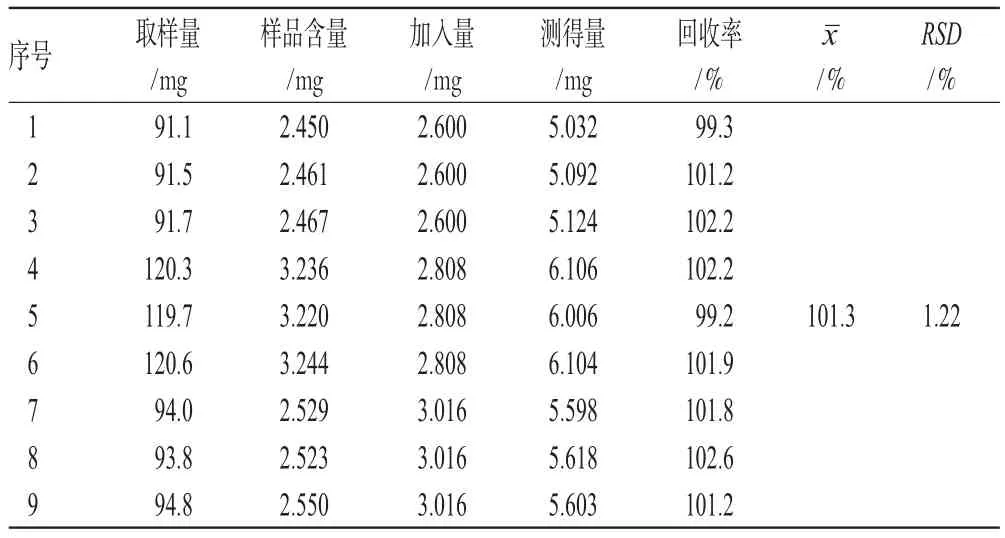
表1 加样回收率试验结果(n=9)Tab 1Results of recovery test(n=9)
2.9 最低检测限测定
将对照品溶液稀释测定,测得信噪比为3∶1时,甘草酸的最低检测浓度为0.083 2 μg·mL-1。
2.10 样品含量测定
取10个不同产地和基原的甘草药材各0.2 g,精密称定,分别按“2.3”项下方法制成供试品溶液,按上述方法测定甘草酸含量,结果见表2。
3 讨论
3.1 流动相的选择
对于甘草酸的含量测定,2005年版《中国药典》采用的流动相为甲醇-0.2 mol·L-1醋酸铵-冰乙酸(67∶33∶1)[1],但预试验却发现甘草酸单铵盐的保留时间太短且峰形不好。经过多次试验,发现以甲醇-0.2 mol·L-1醋酸铵-冰乙酸-三乙胺(64∶36∶1.5∶0.02)为流动相时,样品分离度、对称性及峰形均较好,且保留时间适中,故最终确定本试验的流动相为甲醇-0.2 mol·L-1醋酸铵-冰乙酸-三乙胺(64∶36∶1.5∶0.02)。

表2 10批甘草样品的含量测定结果Tab 2 Content determination of glycyrrhizic acid in 10 batches of samples
3.2 提取溶剂的考察
本研究对提取溶剂(50%甲醇、甲醇、流动相)进行了考察,结果表明用流动相作提取溶剂效率最高,故最终确定用流动相作提取溶剂。
3.3 提取时间的考察
本研究考察了超声提取的时间(20、30、45、60 min),结果表明提取45 min后,提取时间延长并不能显著提高提取效率,故选择提取时间为45 min。
3.4 冷浸时间的考察
本研究考察了冷浸时间(0、10 h)对提取效率的影响,结果表明冷浸对提取效率影响不大,故选择不用冷浸。
3.5 小结
研究显示,不同来源的甘草药材中有效成分甘草酸的含量有较大差异,相差最大的近4倍,其中野生甘草中甘草酸的含量较家种甘草中甘草酸的含量高,编号为8、9、10的3种不同产地的家种乌拉尔甘草的含量低于2%,未达到2005年版《中国药典》的最低标准。可见,不同来源的甘草药材其内在质量存在较大差异,医院在采购药材时应加以注意。
[1]国家药典委员会编.中华人民共和国药典(一部)[S].2005年版.北京:化学工业出版社,2005:59.
[2]慕桂娟.甘草化学成分的研究进展[J].包头医学,2005,29(2):25.
[3]田庆来,官月平,张 波,等.甘草有效成分的药理作用研究进展[J].天然产物研究与开发,2006,18(5):343.
[5]潘英伟,陈卫平,奚丽君.甘草减毒作用的应用与机理分析[J].南京中医药大学学报,2008,24(6):428.
[6]徐淑英,张玉臣.复方甘草酸苷治疗戊型病毒性肝炎疗效观察[J].泰山医学院学报,2008,29(9):715.
Content Determination of Glycyrrhizic Acid inGlycyrrhiza uralensisfrom Different Sources by HPLC
TANG Li-qin,ZHANG Shan-tang,LIU Sheng,SHI Yun-hui(Dept.of Pharmacy,The Affiliated Anhui Provincial Hospital,Anhui Medical University,Hefei 230001,China)
OBJECTIVE:To establish the HPLC method for the content determination of glycyrrhizic acid inGlycyrrhiza uralensisfrom different sources.METHODS:The separation was performed on Hypersil-C18(250 mm×4.6 mm,5 µm)column with mobile phase consisted of methanol-0.2 mol·L-1ammonium acetate-glacial acetic acid-triethylamine(64 ∶36 ∶1.5 ∶0.02)with the flow rate of 1.0 mL·min-1.The detection wavelength was set at 252 nm and column temperature was 30℃.RESULTS:The linear range of glycyrrhizic acid was 0.041 6~0.208 0 mg·mL-1(r=0.999 9)with an average recovery of 101.3%(RSD=1.22%,n=9).CONCLUSION:The content of glycyrrhizic acid is variant in different origin and sources.The content of glycyrrhizic acid in wildG.uralensisis higher than that of domestic species.
Glycyrrhiza uralensis;Glycyrrhizic acid;HPLC;Content;Source
R284.1;R927.2
A
1001-0408(2010)39-3700-02
2009-10-10
2009-11-06)
