乳癖消贴片中标示成分及残留溶剂的含量测定*
2010-08-31霍宁波
霍宁波
(滨州职业学院家纺学院,山东 滨州 256603)
乳癖消贴片中标示成分及残留溶剂的含量测定*
霍宁波
(滨州职业学院家纺学院,山东 滨州 256603)
建立了贴片中标示成分芍药苷含量的HPLC分析方法和贴片中各种残留溶剂含量的GC分析方法。样品经甲醇提取,离心分离后,以乙腈-水(体积比16:84,HAc 0.4%)为流动相,选择ODS-2 Hypersil C18(4.6 mm×150 mm,5 μm)色谱柱,测定芍药苷含量。贴片基质经处理,以GC内标法测定残留溶剂含量。结果表明HPLC法的精密度、重现性和回收率良好;GC分析测得乙酸乙酯、异丙醇、乙醇三种残留溶剂,贴片中残留溶剂总量为2.1×10-4。
乳癖消;压敏胶贴片;芍药苷;残留溶剂;含量测定
乳癖消浸膏由鹿角、蒲公英、昆布等15味中药提取而成,是一种治疗乳腺增生急性乳腺炎等疾病的中药复方。乳癖消贴片是将乳癖消浸膏溶解在丙烯酸酯压敏胶中制得的中药浸膏压敏胶贴片。浸膏中成分复杂,同时压敏胶基质的干扰会影响该贴片中有效成分的测定[1]。水溶性成份芍药苷是乳癖消浸膏中的主要药效成分,含量比较高,便于测定。
文献中关于芍药苷的测定多针对片剂、颗粒制剂等[2-7],目前未见关于HPLC法测定压敏胶贴片中标示成分含量的报道。因此本实验选取芍药苷作为标示成分,建立HPLC法测定乳癖消压敏胶贴片中芍药苷含量,为该压敏胶贴片的质量控制提供参考。
外用制剂中残留的有机溶剂会对皮肤产生刺激作用,引起皮肤过敏反应,中国药典中对外用制剂中残留溶剂的限度(5×10-4)做了规定[8]。丙烯酸酯压敏胶中含有多种溶剂,且基质制备过程中需要加入溶剂,尽管在压敏胶贴片制备过程中绝大部分干燥挥发,但其中可能含有少量溶剂残留,本实验建立了气相色谱内标法测定贴片中有机溶剂的残留量[9-10],以便于该压敏胶贴片的质量控制。
1 实验部分
1.1 仪器与试剂
电子天平;超级恒温水浴;LC-2010A高效液相色谱系统(日本岛津);Angilent 6890气相色谱仪(美国安捷伦);超声仪;高速离心机。
乳癖消压敏胶贴片(自制);芍药苷对照品(中国药品生物制品检定所提供);甲醇、乙腈(均为色谱纯),乙酸乙酯、正庚烷、异丙醇、无水乙醇、苯、乙醚、丙酮(均为分析纯)均购自天津科密欧化学试剂开发中心。HPLC法流动相使用前过0.45 μm微孔滤膜,并超声脱气30 min。
1.2 高效液相色谱条件
ODS-2 Hypersil C18柱(4.6 mm×150 mm,5 μm)色谱柱;柱温:30℃;检测波长230 nm;流动相:乙腈-水(体积比16:84,HAc 0.4%);流速1 mL/min;进样体积10μL。
1.3 气相色谱条件
FID检测器;FFAP柱(30 m×0.53 mm×1 μm);柱温65℃恒温10 min;载气:氮气;流速2 mL/min;分流比50:1;进样口温度250℃;检测器温度250℃。
1.4 样品前处理
1.4.1 芍药苷含量测定样品前处理
取3 cm×3 cm贴片,除去背衬层,精密称定基质重量0.33 g,置100 mL具塞锥形瓶中,加入甲醇5 mL,超声20 min,离心(5 000 r/min)10 min,取上清液备用。沉淀物再加甲醇5 mL,同法操作2次,充分抽提,合并上清液全部转移至25 mL容量瓶甲醇定容,过滤。进样10μL,按外标法以峰面积计算。
1.4.2 残留溶剂含量测定样品前处理
取3 cm×3 cm贴片,除去背衬层,精密称定基质重量0.33 g。乙醚溶解基质后离心(8 000 r/min)10 min,取上层清液置10 mL容量瓶中,加入浓度为0.148 mg/mL的丙酮1 mL作内标物,乙醚定容,作为供试品溶液。
2 结果与讨论
2.1 HPLC分析中样品提取剂的选择
按“1.4.1”的方法,4份乳癖消贴片基质质量分别为0.083、0.082、0.079、0.082 g,分别用70%、80%、90%和100%甲醇-水溶液提取,25 mL容量瓶中定容,分别进样10μL测定峰面积。由表1可见以甲醇提取所得提取率最高。
2.2 HPLC标准曲线的绘制

精密称取一定量的芍药苷置25 mL容量瓶中,加甲醇溶解并定容。用甲醇将其稀释成4.03、8.06、12.10、16.13和32.26 μg/mL的系列标准溶液,分别进样测定。以峰面积(Y)对质量浓度(X,μg/mL)绘制标准曲线。得标准曲线方程:
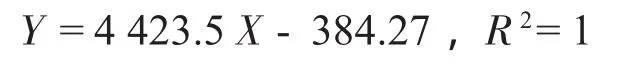
表明芍药苷质量浓度在4.03~32.26μg/mL范围内线性关系良好。芍药苷的HPLC图见图1。

2.3 HPLC分析方法的精密度、重现性与回收率
按含量测定项下制备的同一对照品溶液,连续进样5次,记录峰面积,测得质量浓度分别为21.673、21.555、21.332、21.415、21.154 μg/mL,平均质量浓度为21.426 μg/mL,RSD为0.94%。按含量测定方法取同一批样品分别测定5次,测得样品中芍药苷质量百分含量分别为 0.438%、0.429%、0.432%、0.441%、0.436%,平均含量为0.435%,RSD为1.09%。向5个25 mL容量瓶中分别加入0.5、1.0、1.5、2.0、2.5 mL的已知浓度的芍药苷对照品溶液,分别向其中加入供试品溶液2.0 mL,按供试品测定项下方法操作,测定得芍药苷峰回收率为99.7%~102.8%,RSD为1.17%。
2.4 GC分析中内标物的选择
为了消除手动进样造成的进样体积差异,实验采用内标法测定。根据含有的乙酸乙酯、乙醇等溶剂的沸点、极性以及相互之间有无反应,选择合适的内标物。通过对苯乙酮和丙酮的考察,发现丙酮的保留时间适中且与其它溶剂峰分离度好,适合作为内标物。
2.5 GC色谱条件的选择
可能存在的残留有机溶剂的极性都较小,通过对HP-5柱和FFAP柱的考察,结果表明:HP-5柱出峰慢,受溶剂峰影响明显,且各物质分离度差,这可能是由于该柱为非极性柱的原因;弱极性的FFAP柱对各溶剂的分离效果较好。
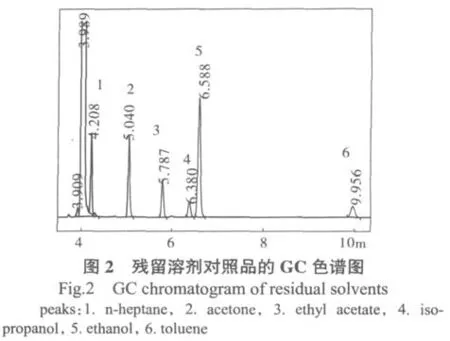
由于各溶剂沸点较为接近,为避免各峰之间的干扰,本试验选择的柱温升温程序为柱温65℃恒温10 min。残留溶剂对照品的GC色谱图见图2。
2.6 GC标准曲线的绘制
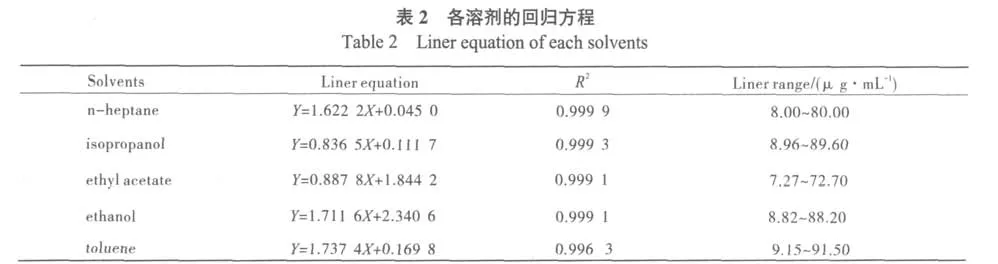
精密称取乙酸乙酯、乙醇、异丙醇、正庚烷、甲苯至100 mL容量瓶中,以乙醚为溶剂,配制浓度分别为0.727、0.882、0.896、0.800、0.915 mg/mL的溶液。精确量取以上5种溶液各0.5、1、2、3、5 mL,分别转移至50 mL容量瓶中,向以上各容量瓶加入0.74 mg/mL的丙酮1 mL作内标,乙醚定容做贮备液。分别进样0.2μL测定。以对照品与内标峰面积比Al/As为纵坐标Y,以乙酸乙酯、乙醇、异丙醇、正庚烷、甲苯与内标物丙酮的质量浓度比Cl/Cs为横坐标X,绘制标准曲线。结果表明:各溶剂线性关系良好,见表2。
2.7 样品测定
气相色谱法测得乙酸乙酯、异丙醇、乙醇三种残留溶剂,未检测到正庚烷、苯,且乳癖消贴片中残留溶剂总量为2.1×10-4(质量分数)。
3 结论
本实验采用HPLC法测定乳癖消贴片中的芍药苷含量,方法操作简单,与杂质峰的分离度好,精密度高,数据准确可靠;采用GC法测定了贴片中的残留溶剂含量,结果符合2005版国家药典小于5×10-4(质量分数)的规定,为这种新开发的中药外用制剂的质量控制提供了参考。
[1]庞谤,杨欣,孙文基.高效液相色谱法测定篇著浸膏片中阿托品的含量[J].药物分析杂志,2008,28(3):414-416.
[2]李捷玮,金柔男,李翔.多种中成药中芍药苷的含量测定[J].药物分析杂志,2009,29(9):1440-1443.
[3]陈春红,韩俊琦,姚永强.HPLC法测定不同溶剂提取的赤芍中芍药苷的含量[J].浙江省医学科学院学报,2006(9):30-31.
[4]谢艳丽.高效液相色谱法测定乳癖消胶囊中芍药苷的含量[J].中国医药导报,2009,6(6):35-36.
[5]姜云云,潘亚菊,范国荣.HPLC法测定乳癖消片中芍药苷含量[J].药学实践杂志,2007,25(5):330-333.
[6]纪松岗,费扬,赵亮.高效液相色谱法同时测定牛黄降压丸中芍药苷和黄芩苷的含量[J].药物分析杂志,2009,29(4):602-605.
[7]谭生建,刘刚,张华.梯度洗脱HPLC法测定抗栓保心片中芍药苷和丹酚酸B的含量[J].药物分析杂志,2009,29(7):110 4-1106.
[8]国家药典委员会.中国药典一部[M].北京:化学工业出版社,1995.
[9]Min J.Yang,Janusz Pawliszyn.Headspace membrane extraction combined with multiplex gas chromatography and mass selective detector formonitoring of volatile organic compounds[J].Journal of Microcolumn Separations,1996,8(2):89-98.
[10]张俊伟.药物中有机残留溶剂的测定方法概述[J].现代仪器,2004(3):10-14.
Determination of Marked Component and Residual Solvents in Rupixiao Patch
HUO Ning-bo
(Department of Textile,Binzhou Vocational College,Shandong Binzhou 256603,China)
A HPLC and GC method was established to detect content of paeoniflorin and residual solvents.After the sample was extracted with methanol and separated by centrifuger,paeoniflorin content was detected with ODS-2 Hypersil C18(4.6 mm×150 mm,5 μm)column,using acetonitrile-water(v/v 16:84,HAc 0.4%)as mobile phase.After the matrix was dissolved by aether and separated by centrifuger,content of residual solvents was detected by using GC internal standard method.Results show that the RSD,reproducibility and recoveries of the method are good;Ethyl acetate,isopropanol and ethanol have been detected by GC,with the totall content of residual solvents 2.1×10-4.
Rupixiao;PSA Patch;Paeoniflorin;Residual Solvents;Determination of Content
0 657
A
1671-0460(2010)01-0102-04
2010-01-09
霍宁波(1980-),男,山东滨州人,助教,从事丙烯酸酯压敏胶的相关研究。E-mail:ningbohuo@yahoo.com.cn。
