化探样品中微量元素 Ag、W、Mo、Sn、Bi、Cu、Pb、Zn、Ni、Co、Cr的水平撒料光谱定量分析
2010-06-06胡道荣
胡道荣
(中南地质勘查院新疆分院,新疆 乌鲁木齐 830063)
地球化学找矿,要求测定化探样品中痕量元素的检出下限达到10-6甚至更低的含量。这类样品通常是大批量的,而且是多元素分析,因此,要求分析方法具有灵敏度高、准确、快速、成本低等特性。
我们所目前分析化探样品所采用的方法有化学法、原子吸收法、极谱法等,虽然这些方法对W、Mo、Sn、Bi等元素可达到所要求的灵敏度和准确度,但是这些方法测定往往是单元素分别进行,所以不但成本高,而且速度慢,分析周期长。再者,Ag在我所现有原子吸收分光光度计上采用火焰法测定的下限只能达到5×10-6,与化探样品中Ag的检出限要求相关甚远,无法满足化探样品的分析。为填补这一空白,根据水平电极撒料法具有快速,灵敏,分馏效应小,对易挥发元素、中等挥发元素甚至某些难挥发元素可以同时测定的特点[1],借鉴其它单位的经验,对原有的W-100平面光栅摄谱仪进行改装,我们在前人工作的基础上做了一些条件试验,并结合本单位的实际情况,拟定了一个区域化探急需的,要求分析灵敏度较高的准确性要符合定量要求的方法。本法采用了特制的水平电极撒样漏斗,改善了下料的均匀性,提高了分析精度;同时选用了SiO2、石墨粉及Na2SO4混合缓冲剂,它能提高弧烧的稳定性,使样品从电极间平缓地进入弧焰,在蒸气云中保持足够的原子及离子浓度,获得了较高的再现性。本法操作简便、快速、成本低,各元素的检出限分别为:Ag 0.03×10-6,W 0.5×10-6,Mo 0.05×10-6,Sn 0.4 ×10-6,Pb 1×10-6,Zn 3×10-6,Cu 0.5×10-6,Bi 0.1×10-6,Ni 0.3×10-6,Co 0.3×10-6,Cr 1×10-6,基本上达到了化探普查找矿定量分析的要求。
1 标准配制
(1)人工基物组分:SiO270%;Al2O315%;Fe2O35%;CaCO32%;MgO 2%;K2SO43%;Na2SO43%。
(2)标准系列:准确称取定量的 Ag、W、Mo、Sn、Bi、Cu、Pb、Zn、Ni、Co、Cr的光谱纯氧化物加入上述人工基物(A)中,使基体中金属含量 Cu、Pb、Zn、W、Sn、Bi、Co、Ni、Cr为 0.3%;Ag、Mo为0.03%,研磨均匀后即得地(1)号标准。
即:(1)号:0.3%Cu、Pb、Zn、W、Sn、Bi、Co、Ni、Cr;0.03%Mo、Ag。
然后按1、3级差用人工基物(A)冲稀到(9)号,得到一套标准系列,详见表1。

表1 标准系列
2 仪器及工作条件
2.1 摄谱条件
摄谱仪:W-100一米平面光栅摄谱仪;光栅刻线1200条/mm,闪耀波长570 nm,二级光谱;狭缝7 μ;三透镜照明系统;中间光栏5 mm×10 mm。
激发光源:WPF-2交流电弧发生器,输入电压220 V,电流强度20 A。
预燃时间:5 s。
曝光时间:16 s(I=20A),下料 300 mg。
电极:直径6 mm左右电极均为锥形头。
感光板:天津I型紫外感光板。
2.2 暗室处理及光度测量
暗室处理:按国产板A、B液及定影液配方配制显影液和定影液,在20℃显影3 min,于定影液中定影至透明,约5~10 min,水冲洗 10 min。
光度测量:东德GFE760 μ测微光度计,P标尺,测量狭缝宽26 μ,高16 mm。
3 水平电极撒料装置的制备
与垂直电极装样法相比,水平电极撒样法除省去车制电极的繁琐外还有着不少的优点[2]:它在燃弧时间内,能保证大量试样均匀地进入弧焰,发射谱线强度大,因而能改善元素的检出限,同时可缩短曝光时间,提高分析速度;此外,它的弧焰较为稳定,且分馏效应小,因此,特别适合于化探样品中的多元素分析。
目前,撒样法最广泛和普遍采用的下样方法是滑槽或传送带下样,滑槽法样品下到电弧中的速度不易控制,而传送带法容易引起样品沾污,而且这两种方法进入弧焰中的样品都不够均匀,再加上下料漏斗口都较大,一般为3~4 mm(如太小则易发生堵塞),这样由于样品粒度、粘性及其他因素影响造成试样沿漏斗壁不同位置下到电弧中的位置便会相应变化而引入偶然误差。对此我们采用了一种特制漏斗,如图1所示。这种漏斗主要是通过振荡器的振荡,使得漏斗上的玻璃棒产生振动及转动,从而使漏斗中的样品能够以一定的速度均匀地进入弧焰,因此它大大改善了下料的均匀性。

图1 水平电极撒样装置

图2 四刻槽平头形插棒
我们发现玻璃插棒头的形状对下料的速度、均匀性及样品沾污程度的影响较大。我们对三角锥形、四角锥形、三槽三角锥形、三槽四角锥形、四槽园锥形、三槽平头形、四槽平头形等不同形状的插棒头进行了对比试验,发现四槽平头形插棒的效果最佳,这种插棒的下料比较均匀,而且还能减少沾污,因此采用了如图2所示的四刻槽平头形插棒。
4 单元素线减光器的制作及应用
本法中的Ag的分析线选择3280.7A和3382.9A,其中,3280.7的测定上限为3×10-6,而3382.9A的测定上限也只能测到5×10-6,为了使Ag5×10-6~30×10-6可以测定而又不降低其他元素分析线的灵敏度,我们采用了单元素、单谱线的减光器,将它固定于暗箱出口处Ag3382.9A谱线的相应位置上,结果证明达到预期目的。
这种减光器的制作:选择透明度好,厚度约0.1 mm的云母片,擦洗干净后放入真空镀膜机去镀一薄层Au薄。其镀膜工艺是[4]:将10~15 mg的金丝或Au薄挂在镀膜机的W丝上,加热使Au熔化沾在W丝上后,停止加热,抽真空,达到真空度后加热喷镀,取出后剪成3×12(mm×mm)的镀Au云母片,贴在做好的架上,置于光谱出口位置上。如图3所示。

图3
5 内标元素的选择
在光谱定量分析中,为了克服工作条件变化对谱线强度带来的影响,常需引入内标元素。
由于撒样法的各元素蒸发特性曲线目前条件下无法实验和绘制,所以内标的选择只能从查阅资料着手,参考垂直电极法的蒸发曲线,结合尝试法来实现。为此我们对易挥发元素选择Ge、In、Sb;对中等挥发及难挥发元素选择Sc、Au、Pd作比较,实验表明,Ge和Pd的效果较为理想,因此选用Ge作Pb、Zn、Ag、Bi、Sn、Mo的内标,用 Pd作 Cu、Ni、Co、W、Cr的内标。
6 缓冲剂的选择及配制
缓冲剂的作用,在多元素分析中很重要,特别是化探样品组分变化复杂,当标准与样品组分不一致时,则产生标准元素黑度值大,样品分析元素黑度值小,使结果偏低的影响,反之则会使结果偏高。因此,在光谱定量分析中往往引入缓冲剂,其目的主要有:消除组分影响;提高被测元素的灵敏度;改善分析方法的再现性等。
由于低电离电位元素(如K、Na等碱金属元素)对电弧温度影响很大,而所分析样品中K和Na的量变化较大,如果在缓冲剂中不加入一定量低电离电位元素,就必然影响测定的稳定性和准确性。据资料[3]介绍,K2SO4能稳定弧焰,改善弧烧状况。而资料[4]又报导,若在缓冲剂中加入NaF,则电弧温度将随NaF量的变化而变化,而当缓冲剂中NaF量为2%~6%时弧温变化较小,为此我们选择了含低电离电位元素的化合物(如KCl、KClO3、NaF、Na2SO4、K2SO4等)作稳定剂,以 SiO2,基体及 C粉等作携带剂和稀释剂,将它们按不同的组合及不同的比例混合,进行试验比较,发现SiO2、C粉中加NaF与SiO2、C粉中加Na2SO4的效果都不错,而且后者效果更好。最后,选择了SiO2∶C∶Na2SO4=61∶30∶9 的混合物作缓冲剂。这种缓冲剂能提高弧烧的稳定性,使样品从电极间平缓地进入弧焰,在蒸气云中保持足够的原子及离子浓度,获得了较高的再现性,同时能使W、Mo、Bi等元素的分析灵敏度稍有提高。
在配制缓冲剂时还加入了内标元素,其组成为:SiO261 g;C(石墨粉)30g;Na2SO49g;GeO20.125g;PdO0.006g。将它们混合磨匀备用。
7 分析手续及分析线对
按样品∶缓冲剂=1∶1的混合比例,分别称取样品(包括被测样品、标准样品、管理样品)150 mg和缓冲剂150 mg,混合磨匀备用。
在前面所述的仪器及工作条件下进行摄谱、洗板和测光,以△P-logC绘制工作曲线,查出含量。
测定所采用的分析线对见表2。
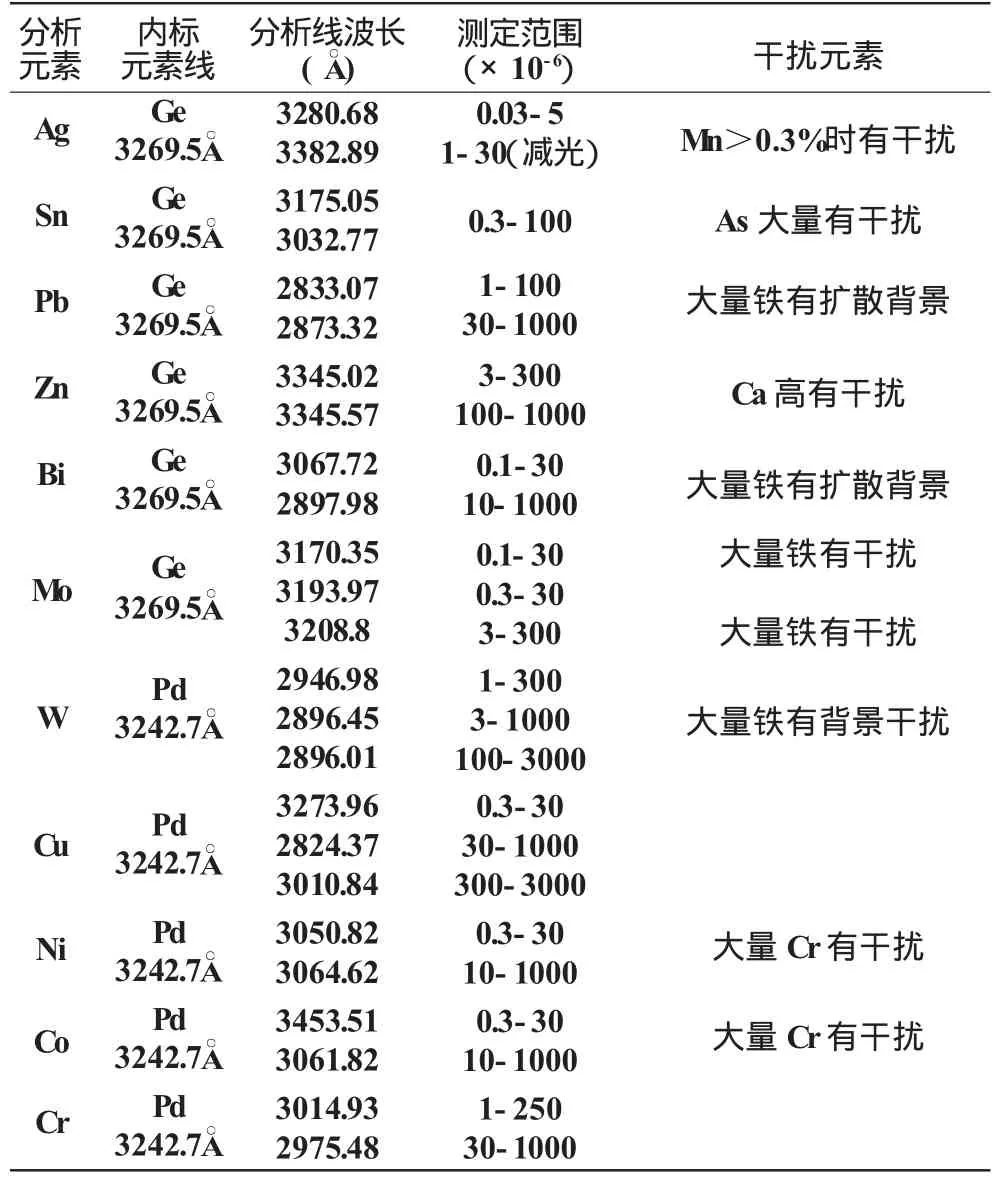
表2 分析线对及测定范围
8 方法的检出限和精密度
8.1 检出限
检出限是衡量分析方法优劣的重要技术指标之一,它通常是指某一分析方法能可靠地检出试样中某元素的最低值。为此,我们取空白样品摄谱20次,按检出限公式:XL=+KS0进行测量计算求出。式中为空白样品测量信号值的平均值,S0为空白样品测量信号值的标准偏差,K为根据选定的置信度所确定的常数值,当K=3时,XL为元素的分析线的检出限信号值,然后以此在标准工作曲线或其延长线上查出的CL值,即为所测元素分析线的检出限。本法测得的各元素的检出限列于表3。
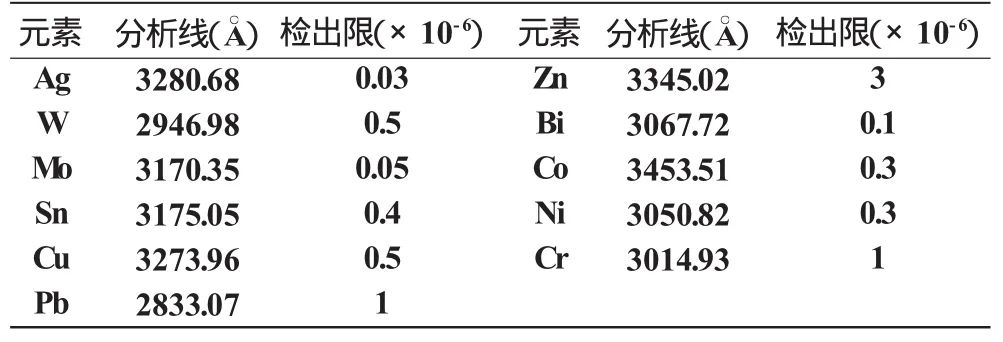
表3 各元素的检出限
8.2 精密度
我们选用了四个综合样品各进行15次摄谱分析,得出各元素的每次含量及15次的平均含量,然后计算出各元素在某含量时的相对标准偏差(RSD%),即为本方法的精密度。见表4。
9 回收试验
在人工配制的基体中加入一定量的被测元素,然后按本法的工作条件进行测定分析,求出含量,并计算回收率。见表5。
10 标样分析结果及对照
用本法对十个地球化学标准样品(GSD、MGD)进行三次分析,各元素分析结果的平均值与标样定值的对照情况列于表6。
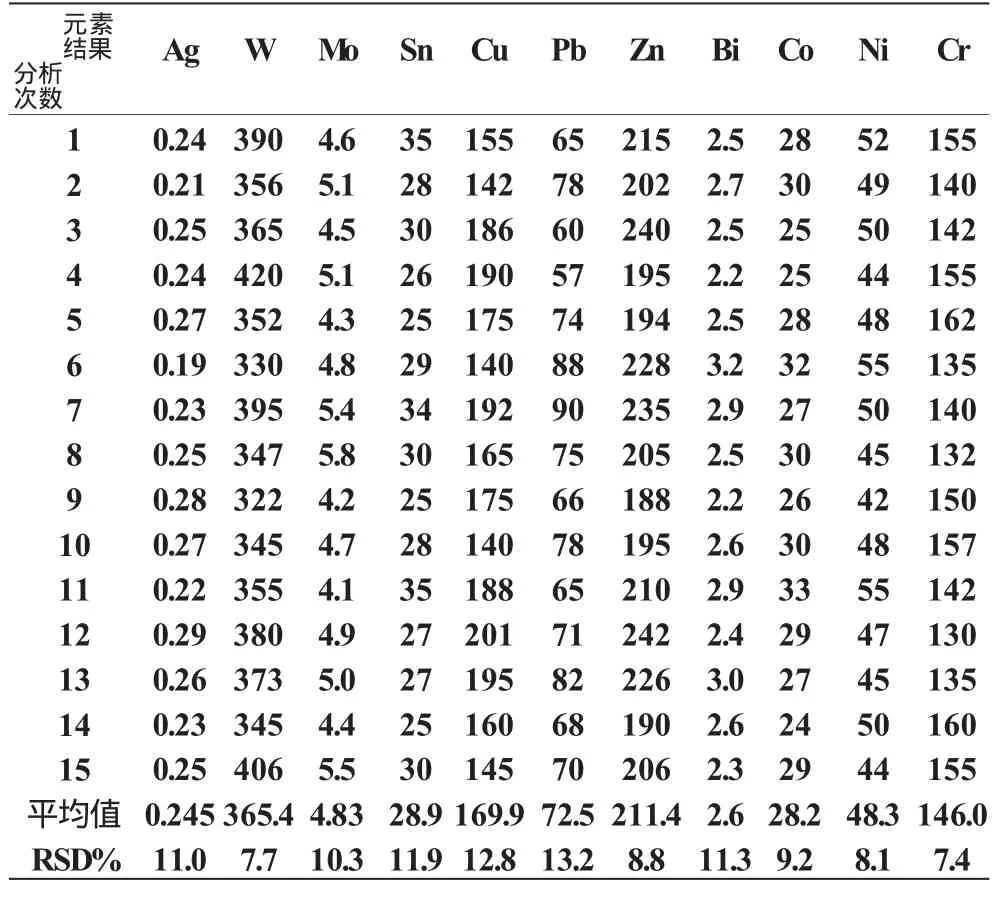
表4 精密度(分析结果单位:W(B)/10-6)

表6 GSD、MGD分析单位:W(B)/10-6
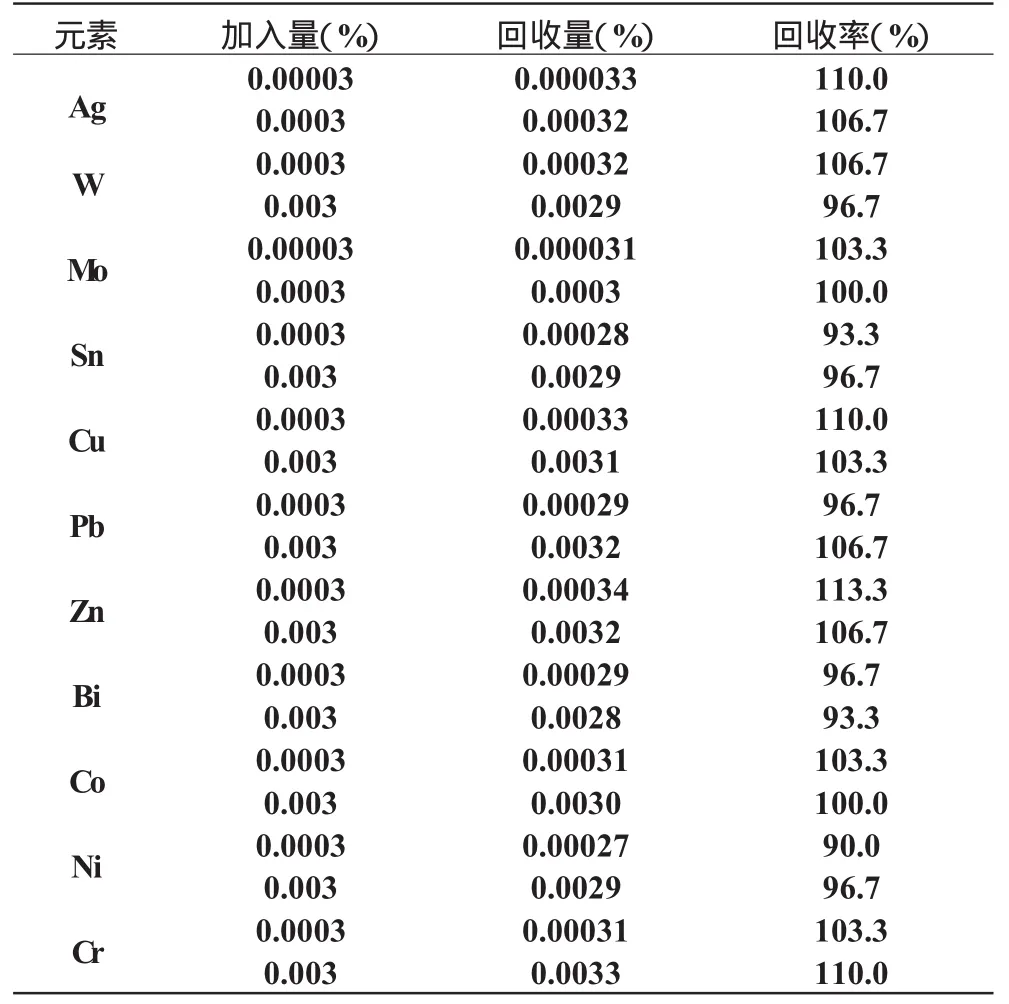
表5 回收率
11 讨论
(1)本法适用于以硅酸盐为主的样品分析,样品中大量的铁、钙等会对 Pb、Zn、Bi、W、Mo等元素产生干扰。
(2)本法灵敏度较高,所分析元素的检出限基本达到或超过中国有色金属总公司地质局九二年下发的《有色地质分析测试质量管理办法》的要求。
(3)本法省去了车制电极的繁琐,操作比较简单,成本较低。
(4)为校正工作曲线,需摄较多的管理样品。
(5)实验表明此法再现性较好,但条件变化引起的误差较大,只要严格控制条件是可以保证精度的。对于化探样品,如操作比较熟练,每个样品只摄一条谱就可以了,对地质样品或不熟练的操作者,可以采取称样品0.3 g和缓冲剂0.3 g,混匀后分两次摄两条谱进行测定。
(6)采用玛瑙乳钵混样,即费时又易引起样品间的玷污,为此,我们将称好的样品与缓冲剂放入特制托盘中的2~3 mL瓷坩埚内,铺上干净白纸,密闭盖严后,置于振荡器中振荡混匀,这样即省时又避免了玷污。另外,为使下样时不易发生沾堵,最好将混好的样品于烘箱中烘干后摄谱或于干燥器中保存备用。
[1]单永健.南岭钨矿赋存层位中的微量 W、Sn、Bi、Mo、Be、Cu、Sb 光谱定量测定[J].地质地球化学,1982,(1).
[2]李廷均.发射光谱分析[M].北京:原子能出版社,1983.
[3]刘诗达.化探样品中微量Sn、Bi、Cd的光谱定量测定,1985.
