高效液相色谱法测定缬沙坦片的含量
2010-06-01廖洪娟
廖洪娟,张 涛
(1.福建省龙岩市第二医院药剂科,福建 龙岩 364000; 2.江苏华源药业有限公司研发部,江苏 泰州 214500)
缬沙坦是一种高选择性Ⅰ型血管紧张素Ⅱ受体(AT1受体)拮抗剂,具有良好的抗高血压作用,可单用或与其他抗高血压药物合用治疗高血压,适用于各种类型的轻、中度高血压,尤其对血管紧张素转换酶抑制剂不能耐受的患者,且无致咳的不良反应[1-2]。目前国内已有多家药厂开始合成生产缬沙坦原料及其制剂产品[3-4],但我国药典尚未收录,故无统一的含量测定方法。笔者参考有关文献[5-8],建立了测定缬沙坦片含量的高效液相色谱(HPLC)法,现报道如下。
1 仪器与试药
岛津高效液相色谱系统(日本岛津公司),包括LC-10ATVP型溶剂输送泵,SPD-10AVP型紫外检测器;N2000色谱软件(浙江大学);BS210S型、BP211D型电子分析天平(赛多利斯公司);SB2000型超声波振荡器。缬沙坦对照品(中国药品生物制品检定所,批号为100651-200401,含量为98.5%);缬沙坦片(江苏常州第四制药厂,批号为 050301,050504,050702);乙腈、磷酸二氢钾、盐酸、氢氧化钠、30%双氧水(色谱纯),注射用水。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Waters Symmetry C18柱(150 mm ×4.6 mm,5 μm);流动相:磷酸盐缓冲液(0.01 mol/L磷酸二氢钾,pH=3.5)-乙腈(20 ∶80),检测波长:230 nm;流速:1.0 mL/min;柱温:40 ℃ ;进样量 20 μL。在此条件下,色谱图见图1。可知,缬沙坦峰与相邻杂质峰分离度大于2.0,理论塔板数按缬沙坦峰计算大于3 000,拖尾因子不超过1.2,表明该系统适用性良好。
2.2 溶液制备
精密称取缬沙坦对照品50.0 mg,置 100 mL量瓶中,用流动相溶解并稀释至刻度,摇匀,作为对照品贮备液;取样品20片,精密称定,研细,取约10.0 mg,精密称定,置100 mL量瓶中,加适量流动相,超声溶解3 min,冷却,加流动相稀释至刻度,过滤,取续滤液作为供试品溶液。取不含缬沙坦的阴性样品,同法制备阴性对照品溶液。
2.3 方法学考察
进样精密度试验:精密移取对照品贮备液2.0 mL至10 mL量瓶中,加流动相稀释至刻度,摇匀,作为对照品溶液,依法连续进样6次。结果峰面积的 RSD为0.48%(n=6),表明进样精密度良好。
中间精密度试验:取批号为050301的样品,委托其他技术人员照供试品溶液制备方法制备溶液,用美国Waters 600高效液相色谱系统依法测定含量。结果平均含量为99.59%,RSD为0.31%(n=6),表明该色谱条件精密度良好。
重现性试验:取批号为050301的样品,依法制备供试品溶液并测定。结果平均含量为99.74%,RSD为0.33%(n=6)。
线性关系考察:分别精密移取对照品贮备液1.0,2.0,3.0,4.0,5.0,6.0 mL,置 10 mL量瓶中,加流动相稀释至刻度,摇匀,分别取20 μL注入液相色谱仪,记录色谱图。以峰面积对溶液质量浓度作线性回归,得回归方程 A=78 145 653 C+254 359,r=0.999 9(n=6)。结果表明,缬沙坦进样质量浓度在0.05~0.3 g/L范围内与峰面积线性关系良好。
定量限与检测限确定:取线性关系考察项下质量浓度为50μg/mL的对照品溶液,继续稀释进样。质量浓度为50 ng/mL时,其基线信噪比约为10∶1,故定量限约为50 ng/mL;继续稀释进样,质量浓度为20 ng/mL时基线信噪比约为3∶1,故检测限约为20 ng/mL。
稳定性试验:取同一供试品溶液,分别于 0,1,2,4,6,8 h 时依法测定。结果峰面积的 RSD为0.25%,杂质峰总峰面积的 RSD为0.98%(n=6),表明供试品溶液在8 h内基本稳定。
加样回收试验:取已知含量的样品,分别按80%,100%,120%的比例精密加入缬沙坦对照品适量,按供试品溶液制备方法制备溶液,依法测定含量,计算回收率。结果见表1。
2.4 样品含量测定
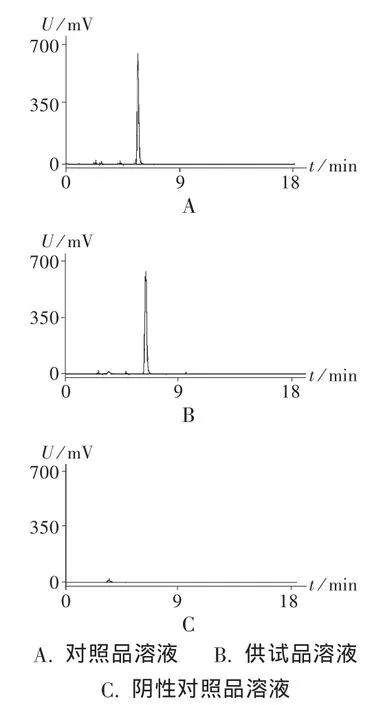
图1 高效液相色谱图
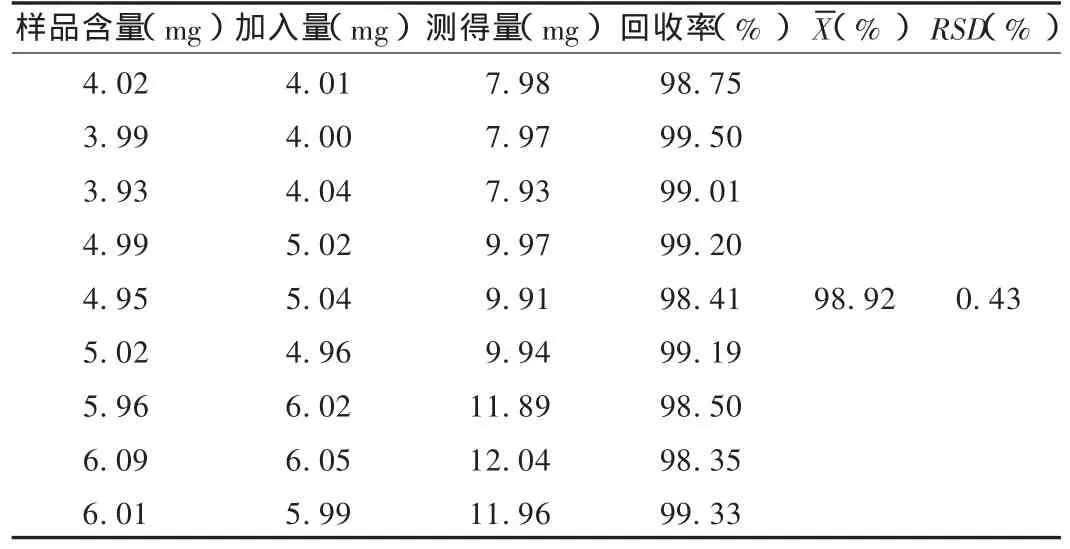
表1 缬沙坦加样回收试验结果(n=9)
精密称取样品适量(约相当于缬沙坦10 mg),置100 mL量瓶中,加入适量流动相,超声溶解3 min,冷却并稀释至刻度,作为供试品溶液;另取缬沙坦对照品适量,同法制成每1 mL含10 μg缬沙坦的溶液,作为对照品溶液。各取20 μL注入液相色谱仪,记录色谱图,按外标法以峰面积计算含量。结果批号为050301,050504,050702的样品中缬沙坦的标示百分含量分别为99.76%,99.28%,99.61%。
3 讨论
有关文献的含量测定方法中,缬沙坦的HPLC法检测波长为270 nm,而其药品试行标准中检测波长为230 nm。在试验过程中,取样品含量测定项下对照品溶液,照分光光度法,于200~400 nm波长范围内测定,结果在230,250,270nm波长处均有吸收,但230nm波长处最大,故检测波长定为230 nm。
在不改变流动相组成成分的条件下,流速加大,主峰的出峰时间会提前;减少流速,主峰的出峰时间会延后,但主峰和其他杂质峰的分离度、理论塔板数、拖尾因子均符合规定。
换用其他型号的C18柱,如Waters Symmetry C18柱(250 mm×4.6 mm,5 μm),Waters Novpak C18柱(150 mm ×3.9 mm,5 μm),Kromasil C18柱(150 mm ×4.6 mm,5 μm),在流动相无改变的情况下,出峰时间仅与色谱柱的长度有关,长柱的出峰时间稍长,短柱出峰时间略快,且主峰和其他杂质峰的分离度、理论塔板数、拖尾因子均符合规定。结果表明,本方法的系统耐用性比较稳定。
[1]杨宝峰.药理学[M].第6版.北京:人民卫生出版社,2006:228-229.
[2]祝世法,顾 亮,王 雁,等.新型血管紧张素受体拮抗剂——缬沙坦[J]. 心脑血管病防治,2001(02):37-39.
[3]曾运才 .缬沙坦的合成工艺研究[J].胶体与聚合物,2003,21(3):43-44.
[4]贾庆忠,马桂林,黎文志,等.抗高血压药缬沙坦的合成[J].中国医药工业杂志,2001,32(9):385.
[5]韩 俊,马宝玉,李 娟.缬沙坦分散片的制备及含量测定[J].时珍国医国药,2007,18(1):145-146.
[6]WS-455(X-391)-2000,国家食品药品监督管理局国家药品试行标准·缬沙坦[S].
[7]WS1-(X-2339)-2003Z,国家食品药品监督管理局国家药品试行标准·缬沙坦[S].
[8]国家药典委员会.中华人民共和国药典(二部)[M].北京:化学工业出版社,2005:附录 172,附录 182.
