清脑宁颗粒质量标准研究*
2010-06-01李福如顾海琳洪雪勤
李福如,缪 婧,顾海琳,洪雪勤
(1.江苏省南通市中医院,江苏 南通 226001; 2.南通大学,江苏 南通 226001)
清脑宁颗粒是我院脑外科的科研制剂,由赤芍、当归、川芎、陈皮等10味中药组成,具有活血化瘀、镇痛、安神的功效,对脑外伤后的头痛、失眠及脑功能恢复有较好疗效。根据处方药物的理化性质,笔者采用薄层色谱(TLC)法对方中主药(赤芍、当归、川芎、陈皮)进行定性鉴别,采用高效液相色谱(HPLC)法对赤芍中芍药苷进行含量测定,报道如下。
1 仪器与试药
岛津LC-20AD型高效液相色谱仪。层析用硅胶(青岛海洋化工厂);芍药苷对照品(批号为110736-200731)、阿魏酸对照品(批号为110773-200611)、橙皮苷对照品(批号为110721-200613),均购自中国药品生物制品检定所;甲醇为色谱纯,其他试剂均为分析纯。
2 方法与结果
2.1 TLC鉴别
赤芍[1]109:取清脑宁颗粒1 g,加乙醇20 mL,振摇5 min,滤过,滤液蒸干,残渣加乙醇2 mL使溶解,作为供试品溶液;按处方制备不含赤芍的阴性样品,同法制备阴性对照品溶液;取赤芍对照药材1 g,同法制成对照药材溶液;另取芍药苷对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。吸取上述4种溶液各5 μL,分别点于同一硅胶G薄层板上,以氯仿-醋酸乙酯-甲醇-甲酸(40∶5∶10∶0.2)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰,置日光下检视。供试品溶液色谱中,在与对照品药材溶液及对照品溶液色谱相应位置上显相同颜色的荧光斑点,而阴性对照品溶液色谱中则无此斑点(图1 A)。
当归[1]89:取清脑宁颗粒4 g,加1%碳酸氢钠溶液50 mL,用力搅拌,滤过,滤液用稀盐酸调节pH至2~3,用乙醚10 mL振摇提取,取上层乙醚液,置水浴挥干,残渣加甲醇1 mL使溶解,作为供试品溶液;按处方制备不含当归的阴性样品,同法制备阴性对照品溶液;取当归对照药材1 g,同法制成对照药材溶液;另取阿魏酸对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。吸取上述4种溶液各3 μL,分别点于同一硅胶G薄层板上,以苯-醋酸乙酯 -甲酸(4∶1∶0.1)为展开剂,展开,取出,晾干,喷以新配制的1%三氯化铁和1%铁氰化钾(1∶1)的混合溶液,置日光下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的荧光斑点,而阴性对照品溶液色谱中则无此斑点(图1 B)。
川芎[1]28:取当归鉴别项下供试品溶液,作为供试品溶液;按处方制备不含川芎的阴性样品,同法制备阴性对照品溶液;取川芎对照药材1 g,同法制成对照药材溶液;另取阿魏酸对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。吸取上述4种溶液各5 μL,分别点于同一硅胶G薄层板上,以苯-醋酸乙酯-甲酸(4∶1∶0.1)为展开剂,展开,取出,晾干,喷以新配制的1%三氯化铁和1%铁氰化钾(1∶1)的混合溶液,置日光下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的荧光斑点,而阴性对照品溶液色谱中则无此斑点(图1 C)。
陈皮[1]132:取清脑宁颗粒 0.5 g,加甲醇 10 mL,振摇 5 min,滤过,滤液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液;按处方制备不含陈皮的阴性样品,同法制备阴性对照品溶液;取陈皮对照药材1 g,同法制成对照药材溶液;另取橙皮苷对照品,加甲醇制成饱和溶液,作为对照品溶液。吸取上述供试品溶液及阴性对照品溶液2~5 μL,对照药材溶液及对照品溶液2 mL,分别点于用0.5%氢氧化钠溶液制成的同一硅胶G薄层板上,先以醋酸乙酯-甲醇-水(100∶17∶13),再以甲苯 -醋酸乙酯 -甲酸 -水(20∶10∶1∶1)的上层液为展开剂,展开,取出,晾干,喷以三氯化铝试剂,热风吹干后,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液及对照品溶液色谱相应位置上显相同的荧光斑点,而阴性对照品溶液色谱中则无此斑点(图1 D)。
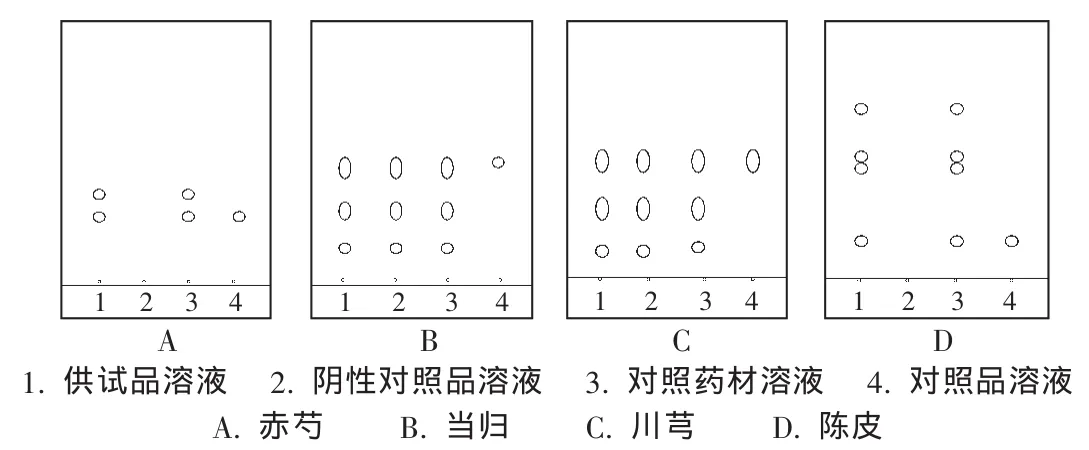
图1 薄层色谱图
2.2 含量测定[1]109,[2-3]
2.2.1 色谱条件与系统适用性试验
色谱柱:岛津 C18柱(250 mm ×4.6 mm,5 μm);流动相:甲醇 -0.05 mol/L 磷酸二氢钾(30∶70);检测波长:230 nm;柱温:25℃;流速:1.0 mL/min;进样量:50 μL。理论塔板数按芍药苷计算应不低于3 000,方中其他成分对芍药苷的含量测定无影响(图2)。
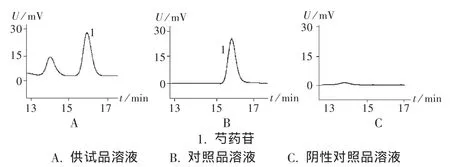
图2 高效液相色谱图
2.2.2 溶液制备
精密称取芍药苷对照品12 mg,置100 mL量瓶中,加流动相溶解并稀释至刻度,摇匀,即得质量浓度为120 μg/mL的对照品贮备液。精密量取对照品贮备液5 mL,置10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得对照品溶液(每1 mL含芍药苷60 μg)。取装量差异项下样品内容物研细,取8 g,精密称定,加入甲醇50 mL,称定质量,超声处理50 min,放置室温,用甲醇补足减失的质量,摇匀,滤过,弃去初滤液,即得供试品贮备液。精密量取供试品贮备液5 mL,置10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得供试品溶液。按处方制备不含赤芍的颗粒,精密称取该空白样品8 g,同法操作制备阴性对照品溶液。
2.2.3 方法学考察
线性关系考察:分别吸取对照品贮备液 1,2,3,5,8 mL,置 10 mL量瓶中,制得质量浓度为 12,24,36,60,96,120 μg/mL 的对照品溶液,分别进样2次,每次50 μL,按2.2.1项下色谱条件测定,记录峰面积,以对照品进样量(X,μg)为横坐标、峰面积平均值(Y)为纵坐标绘制标准曲线,得回归方程 Y=13 924X-195.23,r2=0.999 8(n=6)。结果表明,芍药苷质量浓度在 12~120 μg/mL范围内与峰面积线性关系良好。
精密度试验:取对照品溶液,重复进样测定。结果峰面积的RSD 为 0.67% (n=6)。
稳定性试验:取同一供试品溶液,于室温放置 0,2,4,6,8 h 时进样测定。结果峰面积的 RSD=0.65%(n=5),表明供试品溶液在8 h内稳定。
重现性试验:精密称取样品8.0 g,依法制备供试品溶液并测定。结果含量的 RSD=1.60%(n=5)。
加样回收试验:精密称取已知含量的样品(批号为20080308,含量为7.4 mg/袋,平均装量为10.152 1 g),精密加入0.1 mg的芍药苷对照品,按供试品溶液制备方法制备溶液并进样测定,计算回收率。结果见表1。
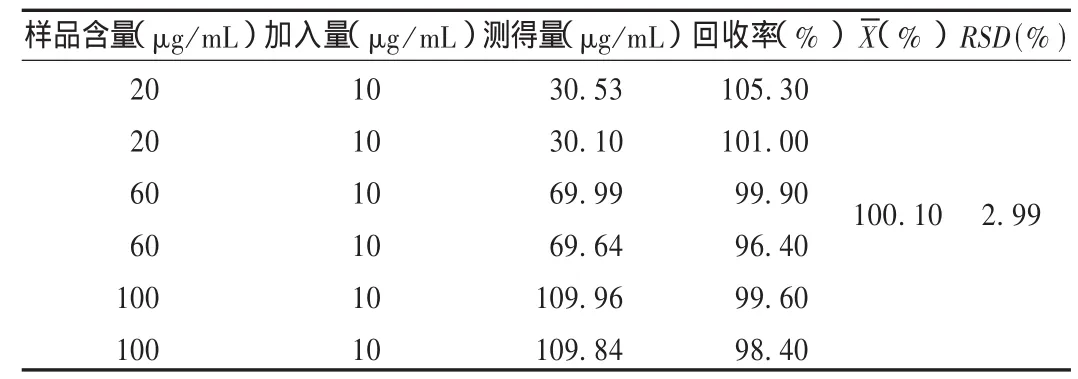
表1 芍药苷加样回收试验结果(n=6)
2.2.4 样品含量测定
取3批样品,依法制备供试品溶液并测定含量。结果批号为080313,080320,080327的每袋样品中芍药苷含量分别为7.11,7.24,7.05 mg,RSD=1.36%。
3 讨论
在TLC色谱中,供试品、对照药材溶液及对照品溶液在相应的位置呈相同颜色斑点,说明本定性鉴别方法是合理可行的。
参照2005年版《中国药典(一部)》赤芍项下含量测定方法中流动相的比例,分析时间较短,但分离度达不到要求,经调整流动相比例,以甲醇-0.05 mol/L磷酸二氢钾(30∶70)为流动相时效果较好。该方法准确度高,加样回收率、重现性好,可作为清脑宁颗粒的质量控制方法。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005.
[2]张 玲.高效液相色谱法测定参梅养胃颗粒中芍药苷的含量[J].药学与临床研究,2008,16(1):77-78.
[3]李道明,周修森.SPE-HPLC法测定补中益肾丸中芍药苷的含量[J].药学与临床研究,2008,16(1):71-73.
