方解石去除水体中高浓度磷酸盐的反应动力学
2010-05-31钟山王里奥刘元元董靖蒙
钟山,王里奥, ,刘元元,董靖蒙
(1. 重庆大学 资源及环境科学学院,重庆,400044;
2. 重庆大学 三峡库区生态环境教育部重点实验室,重庆,400044)
磷污染是导致水体富营养化的主要原因之一,人们对水体中磷的去除进行了大量研究[1-6]。实际工业的含磷废水中磷浓度很高,如涂装废水中磷酸盐质量浓度达到100 mg/L左右[7-8],而磷肥废水中磷酸盐质量浓度更高[9]。方解石属天然矿物,容易获得且成本低廉。已有研究表明,方解石可有效去除水中的磷酸盐[10-11]。Kostantinos等[12]认为碱性条件适宜方解石除磷,吸附作用是方解石去除低质量浓度磷酸盐的主要机制;Song等[13]则认为方解石表面附着磷酸钙的晶体生长是除磷的主要机制;许虹等[14]的实验结果表明温度对方解石除磷效果影响十分显著;林建伟等[15]的研究表明:在常温下方解石对水体中质量浓度为20 mg/L的磷酸盐的去除率可达到98%。目前,相关研究主要集中在方解石除磷的效果、影响因素与反应机理等几方面且多以处理较低浓度磷酸盐为主,对于高质量浓度磷酸盐的处理及动力学方面的研究较少。因此,在处理实际工业废水时,对于反应条件的选择尤其在反应温度与时间控制方面缺乏理论指导。为此,本文作者通过X线衍射分析(XRD)、红外光谱分析(FT-IR)、透射电子显微镜(TEM)检测的手段结合水化学理论研究方解石除磷产物与主要控制步骤,并进一步分析主要控制步骤的反应过程与动力学模型,以便为含磷废水的处理提供科学的理论支撑。
1 材料与方法
1.1 试验材料
(1) 方解石粉末,购自浙江长兴南方微粉厂,粒径为48 μm,碳酸钙质量分数>99%。
(2) 高浓度磷酸盐废水,采用自行配制的质量浓度为100 mg/L的KH2PO4溶液。
1.2 试验方法
1.2.1 基础试验
将100 mL质量浓度为100 mg/L的KH2PO4溶液加入碘量瓶中,用 NaOH 或 HCl 调节 pH(范围为2.00~12.00)后,盖紧瓶塞放入 SHY22 恒温振荡器中(温度为293,303,313,323和333 K)。然后,加入0.500 0 g方解石样品,盖紧瓶塞后以180 r/min 的转速恒温振荡,反应一定时间后离心分离,转速为3 000 r/min。取上清液测定溶液的pH和磷酸盐浓度;将固体残余物反复清洗后离心分离,将沉降物干燥,碾磨成待测样进行 X线衍射分析(XRD)、红外光谱分析(FT-IR)、透射电子显微镜(TEM)检测。
1.2.2 动力学试验
方解石除磷过程中,中间产物CaHPO4·2H2O (即DCPD)转化为 Ca3(PO4)2·xH2O(即 ACP)是主要控制步骤,本文动力学试验主要针对该步骤进行设计。将2.000 0 g CaHPO4·2H2O(DCPD) 投加到装有 200 mL ρ(Ca2+)=0.120 0 mg/L溶液的锥形甁中,放入SHY22恒温振荡器(温度序列为288,298,308和318 K),以转速为180 r/min恒温振荡,监测并保持溶液pH为8.0。在反应一定时间后取出离心分离,取固体残余物反复清洗后再离心分离,将沉降物干燥并测定其钙、磷含量,计算 DCDP转化率。定义 DCPD转化率 X=(m0-m1)/m0(式中:m0为DCPD初始质量;m1为DCPD残余质量)。
1.2.3 主要测定方法
磷酸盐质量浓度采用钼锑抗比色法测定,仪器为722型分光光度计;pH用PHS-3CW 型pH计测定;固相物质中钙含量用EDTA滴定法测定。
XRD检测条件:铜靶,测试电压为40 kV,电流为100.0 mA,扫描速度为2 (°)/min,仪器型号为BDX 3200。
IR检测条件:KBr压片制样,仪器型号为Nicolet 5DXC FT-IR。
TEM检测条件:仪器型号为TECNAI 10,测试电压为100 kV。
2 结果与讨论
2.1 化学沉淀过程
不同处理时间的固相物质 X线衍射谱如图 1所示。从图1可见:反应过程中方解石特征峰强度不断减弱;0.5 h后出现明显CaHPO4·2H2O(DCPD)衍射峰,1 h后该峰达到最高,之后开始减弱并在8 h后消失。表明在0.5 h首先生成的产物有DCPD,这也与文献[14]中的结果相符,1 h生成 DCPD的反应结束。DCPD并不稳定,8 h后大部分转化为其他物质,但XRD谱显示没有其他晶态物质生成,这说明可能有非晶态物质生成。为验证此推测,对产物进行升温转晶,发现β-磷酸三钙(β-TCP)特征峰,同时,方解石特征峰强度减弱,并有Ca(OH)2衍射峰出现,说明在升温过程中,有非晶态磷酸三钙 Ca3(PO4)2·xH2O(ACP)转化为β-TCP,CaCO3分解产生的CaO吸水转变为Ca(OH)2。
不同处理时间的固相物质红外光谱如图2所示。可见:反应前在波长1 427 cm-1和876 cm-1处的峰为CO32-特征吸收峰,并且在整个反应过程中基本不变,其原因是方解石过量投加使固相物质始终以CaCO3为主;反应0.5 h后在1 036 cm-1处出现新的吸收峰,而HPO42-与 PO43-的特征吸收峰均靠近该波频,难以区分,因此,推测固相中HPO42-与PO43-两者都可能含有。随着反应的进行,在1 036 cm-1处的特征峰增强,说明固相中HPO42-与PO43-含量有所增加,同时并没有其他特征基团。结合 XRD检测结果可知反应过程是:先生成DCPD;然后,DCPD转化为ACP。

图1 处理时间不同时固相物质X线衍射谱Fig.1 XRD patterns of CaCO3 and products for different reaction time

图2 不同处理时间的固相物质红外光谱Fig.2 FT-IR patterns of CaCO3 and products for different reaction time
由于磷酸在溶液中存在三级电离,在不同 pH下呈不同形态。在除磷过程中,pH为4.50~8.41,此时,溶液中磷的形态为 H2和,基本上没有PO43-,因此,直接生成 Ca3(PO4)2是难以实现的。可见:在除磷过程中应是先生成DCPD,再向ACP转化,而方解石可为反应提供钙源。可见除磷主要包括2个过程:生成DCPD,DCPD转化为ACP。化学反应式可表示为:
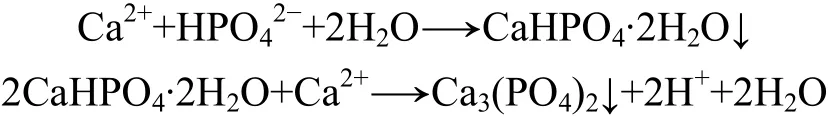
2.2 反应时间与反应温度对方解石除磷效果的影响
处理时间对磷酸盐去除率的影响见图 3。从图 3可以看出:反应进行1 h内,方解石对磷酸盐的去除率达到68%并且反应速率很快;反应2 h后,反应速率明显降低,8 h后去除率达到98%。结合化学沉淀过程的分析可知:生成DCPD的过程速度较快,在温度为313 K时反应1 h即基本完成;DCPD→ACP的平均反应速率要慢得多,完全转化需要8 h以上,这也与XRD检测结果相符。由此表明从DCPD向ACP的转化过程是磷酸盐高去除率的主要控制步骤,并且转化速率随时间的增加而越来越小。
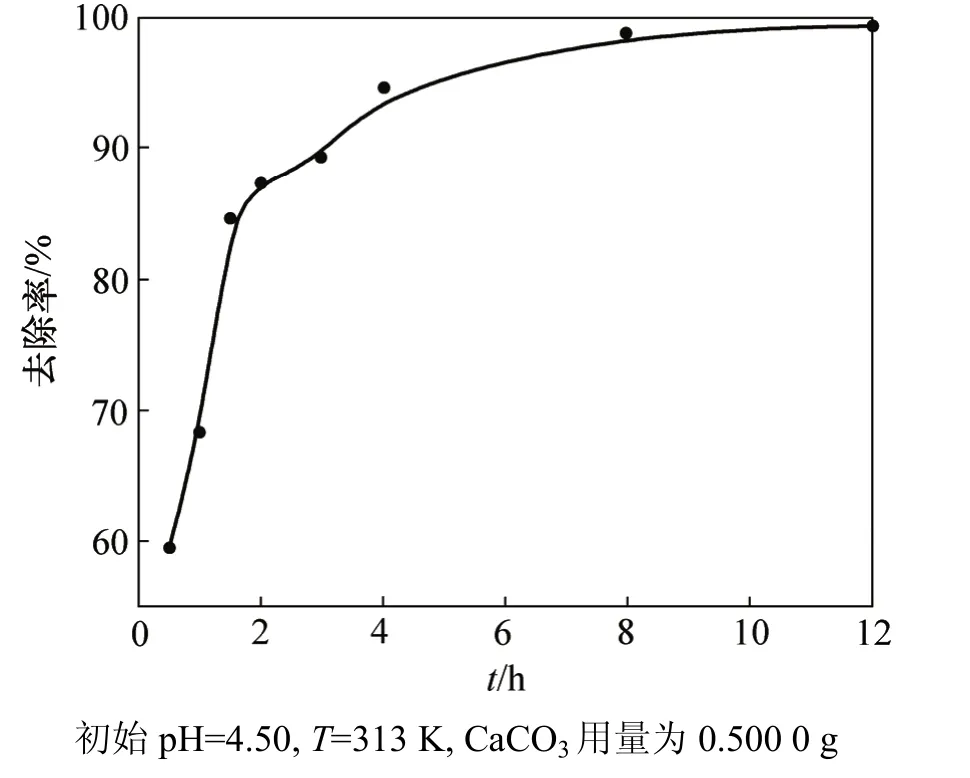
图3 处理时间不同时磷酸盐的去除率Fig.3 Removal of phosphate at different reaction time
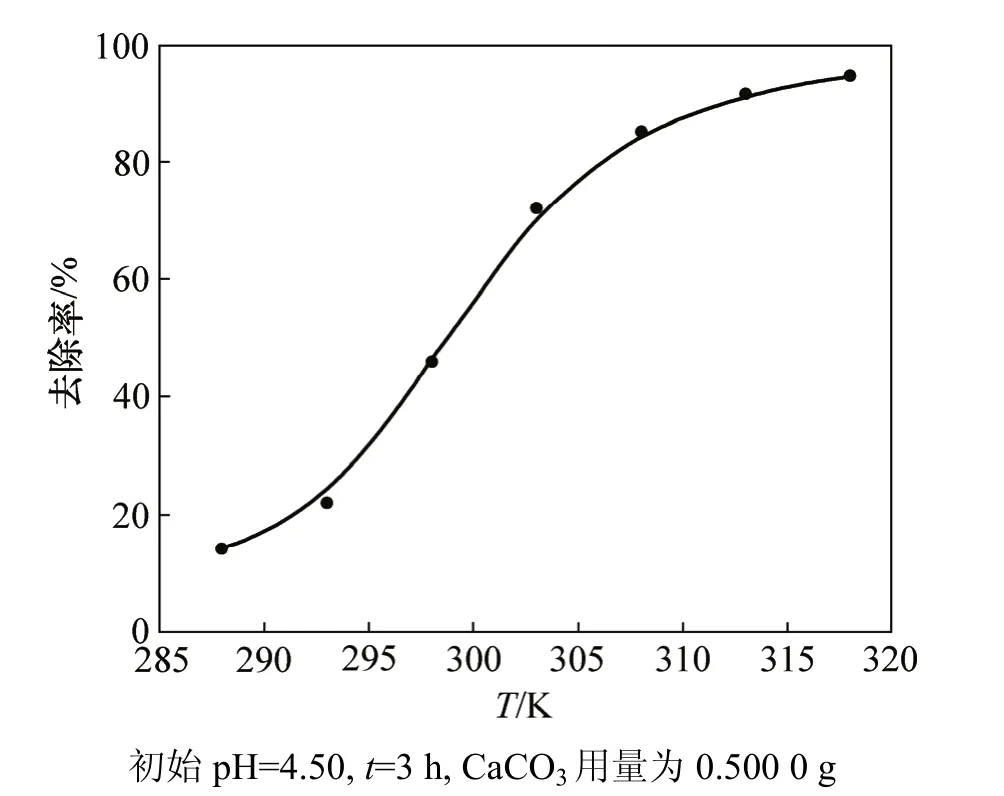
图4 处理温度不同时磷酸盐的去除率Fig.4 Removal rate of phosphate at different temperatures
处理温度对磷酸盐去除率的影响见图 4。从图 4可以看出:当反应温度为288 K时,方解石对磷酸盐的去除率很低,低于20%;当温度升至323 K时,在同样时间内去除率高达98.3%。随着反应温度的增加,磷的去除率显著增加,这表明方解石的除磷过程宏观表现为吸热过程,由此推测作为最主要控制步骤的DCPD向ACP的转化过程也应该是吸热过程。
2.3 ACP生成动力学
2.3.1 动力学模型拟合
DCPD→ACP的转化过程是磷酸盐高去除率的主要控制步骤。利用反应温度对反应速率的影响可以确定 DCPD→ACP的动力学模型。pH监测结果表明反应中溶液的pH基本维持在8.0左右,因此,选择研究pH=8.0时该反应的动力学过程。DCPD→ACP的转化过程是固液反应,属于非均相反应。反应的速率取决于阳离子扩散速率和化学反应速度。由于反应的离子扩散机制和固体颗粒形态都尚未明确,所以,尝试用模型拟合并结合反应过程分析与透射电镜观测的方法研究其动力学过程,选择最为常见的8种动力学模型[16]进行拟合。要进一步判定哪种模型能最好地描述这一过程,必须考虑表观活化能是否符合对应模型的基本要求。根据不同模型拟合出不同温度下的反应速率常数k,采用Arrhenius公式:

其中:k为反应速率常数;k0为指前因子;Ea为表观活化能;R为摩尔气体常数;T为热力学温度。按ln k对1/T作图即可求出表观活化能。一般来说,表观活化能大于42.0 kJ/mol的反应为化学反应控制,反之为扩散控制[16]。也就是说,反应控制型模型拟合结果中表观活化能不应小于42.0 kJ/mol,而在扩散控制模型中,不应大于42.0 kJ/mol。
动力学拟合常数与表观活化能见表1。从表1可以看出:不同模型拟合计算的表观活化能相差较大,而且零级、一级、二级反应模型与界面-反应控制模型的表观活化能也不尽合理,界面-扩散控制模型、平板内扩散控制模型、圆柱内扩散控制模型、球体内扩散控制模型与外扩散模型的表观活化能符合判定标准。模型拟合结果表明扩散控制模型比较适用于DCPD→ACP的转化过程。具体的界面扩散与内、外扩散模型类型还要依据反应本身的实际过程来判断,反应中颗粒形貌则通过透射电镜观察来确定。
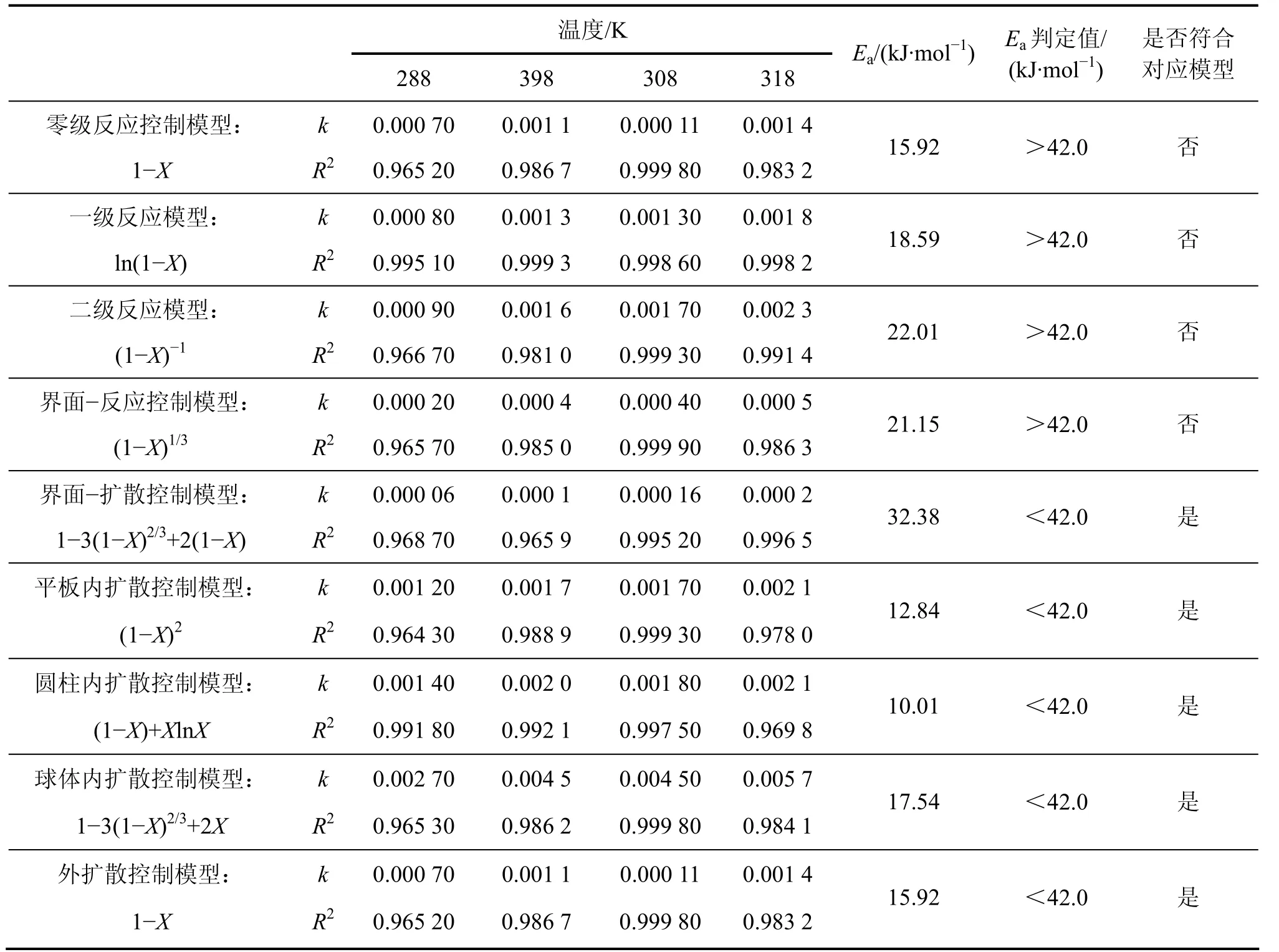
表1 采用不同模型时的动力学拟合常数与表观活化能Table 1 Kinetics constants and apparent activation energy obtained with different models
2.3.2 固相物质透射电镜检测
反应前、后固相物质的TEM照片见图5,DCPD转化率与时间的关系见图 6。从图 5(a)可见:DCPD微观形貌呈板状堆叠形态,每一晶板大小不均,但边缘整齐,轮廓清晰,晶体表面平整,说明其晶体结构完好;在其右上可见部分较小的板状晶体堆积,厚度大,形成较大衬度。从图5(b)可见:DCPD晶体边缘参差不齐,轮廓开始变得模糊,晶体表面粗糙不平,但整体板状形貌仍较清晰。由此说明在DCPD表面有反应发生,这一反应是DCPD→ACP转化,由表面向内部推进,因此,DCPD表面形貌变化较明显,此时,DCPD转化率约为27%(见图6)。虽然表面形貌有所改变,但其整体结构变化不大。从图 5(c)可见:DCPD晶体表面粗糙,边缘轮廓模糊,并伴有小颗粒晶片脱落,但总体仍基本维持层状形貌,此时,DCPD转化率已经接近60%,在晶体表面和边缘反应进行得比较充分,形貌变化比较明显。由于ACP是附着在DCPD晶体表面生成的,大颗粒晶层总体上还保留着DCPD的假象形貌,因此,在DCPD→ACP转化的过程中,固相物质虽然表面形貌发生一些改变,但整体仍基本维持板状形貌。所以,在动力学模型中,微观颗粒形貌宜假定为平板状。
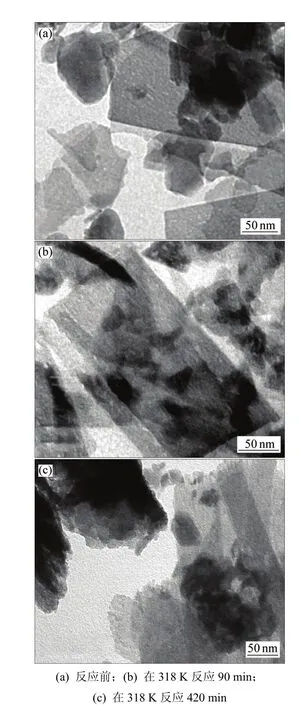
图5 反应前、后固相物质TEM照片Fig.5 TEM photographs of DCPD before and after reaction
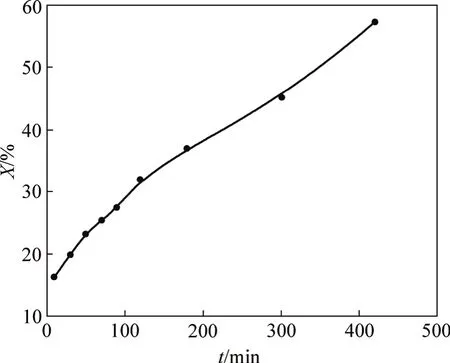
图6 DCPD转化率X与时间t的关系Fig.6 Relationship between transformation rate of DCPD reaction time
2.3.3 反应过程理论模型
固液相反应过程中,在DCDP固体颗粒表面很快有反应产物ACP生成并形成固相膜。实际的传质过程包括以下7步:
(1) 反应物Ca2+由液相主体扩散到达固液界面层;
(2) 反应物Ca2+由固液界面层扩散到达ACP固相膜表面;
(3) 反应物Ca2+通过ACP固态膜扩散到未反应的DCPD层表面;
(4) Ca2+与DCPD在ACP与DCPD界面上反应生成ACP与H+;
(5) 生成物H+由DCPD层表面通过ACP固相膜扩散到颗粒外表面;
(6) 生成物H+由颗粒外表面扩散通过固液界面层;
(7) 生成物H+由固液界面层扩散进入液相主体。
由于反应主要发生在DCPD层表面,所以,传质的阻力主要集中在内扩散过程(3)与(5),而外扩散过程(1)与(7)及界面扩散过程(2)与(6)的阻力要小得多,这就是说,DCPD→ACP的转化过程应属于内扩散控制。由于反应的发生使未反应的DCPD表面同时向颗粒中心收缩(见图8),是典型的扩散边界移动的不稳定内扩散过程,其扩散过程与固体颗粒的几何形状有关[17]。
TEM照片显示微观颗粒形貌为平板状,因此,平板内扩散控制模型最适用于描述DCPD→ACP的转化过程。DCPD转化率与时间的关系可以描述为(1-X)2=-k0exp[-Ea/(RT)]t。平板内扩散模拟结果见图7,DCDP→ACP的转化过程理论模型示意图(内扩散)见图8。从图7可见:在本实验条件下,得到指前因子k0=0.271 2,表观活化能Ea约为12.84 kJ/mol。在实验的4个温度条件下(288, 298, 308和318 K)该模型拟合效果都比较好,R2均大于0.96 (见图7)。

图7 平板内扩散模型拟合结果Fig.7 Fitted results of flat plate controlled by internal diffusion model
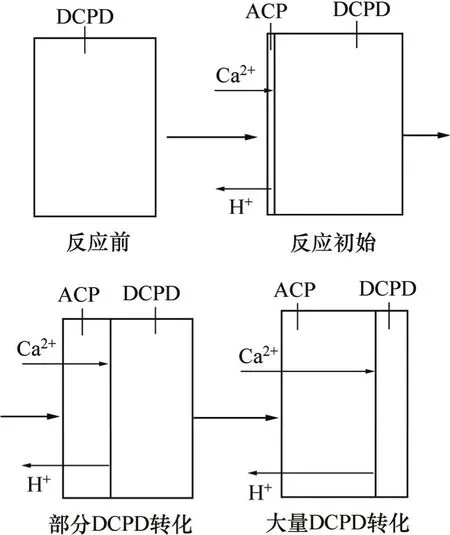
图8 DCDP→ACP的转化过程理论模型示意图(内扩散)Fig.8 Theoretic model of DCDP→ACP
3 结论
(1) 方解石除磷的核心过程包括生成CaHPO4·2H2O(DCDP)沉 淀 与 DCDP 转 化 为Ca3(PO4)2·xH2O(ACP)沉淀,化学反应式可表示为:

(2) DCPD→ACP是方解石除磷达到高去除率的主要控制步骤,属吸热反应;阳离子内扩散过程是影响反应速率的主要因素,该过程适用于平板内扩散控制模型。DCPD转化率与时间的关系可描述为:(1-X)2=-k0exp[-Ea/(RT)]t。在本实验条件下,得到的指前因子k0=0.271 2,表观活化能Ea约为12.84 kJ/mol。
(3) DCPD在反应过程中固相物质颗粒边缘轮廓逐渐模糊,表面变得粗糙不平,但总体仍维持板状形貌,这为平板内扩散的动力学模型的建立提供了有力的微观尺度证据。
[1] Balmer P. Phosphorus recovery—An overview of potentials and possibilities[J]. Water Science and Technology, 2004, 49(10):185-190.
[2] Stratful I, Scrimshaw M D, Lester J N. Conditions influencing the precipitation of magnesium ammonium phosphate[J]. Water Research, 2001, 35(17): 4191-4199.
[3] Lee C W, Kwqn H B, Jeon H P, et al. A new recycling material for removing phosphorus from water[J]. Journal of Cleaner Production, 2009, 19(7): 683-687.
[4] Ago K, Christina V, Riho M. Hydrated calcareous oil-shale ash as potential filter media for phosphorus removal in constructed wetlands[J]. Water Research, 2008, 42(4/5): 1315-1323.
[5] Hosni K, Moussa S B, Chachi A, et al. The removal of PO43-by calcium hydroxide from synthetic wastewater: Optimization of the operating conditions[J]. Desalination, 2008, 223(1/2/3):337-343.
[6] Song Y H, Hahn H H, Hofmann E. Effects of solution condition on the precipitation of phosphate for recovery: A thermodynamic evaluation[J]. Chemosphere, 2002, 48(10): 1029-1034.
[7] 李长江, 郭一令, 王希辉, 等. 高浓度含磷废水治理工艺研究[J]. 环境科学与管理, 2005, 30(5): 61-63.LI Chang-jiang, GUO Yi-ling, WANG Xi-hui, et al. Study on treatment of high concentrated phosphorus containing wastewater[J]. Environmental Science and Management, 2005,30(5): 61-63.
[8] 张显忠, 张智, 魏虎兵. 酸洗磷化废水处理工程[J]. 水处理技术, 2007, 33(8): 85-87.ZHANG Xian-zhong, ZHANG Zhi, WEI Hu-bing. Process and practice of phosphorus containing wastewater treatment[J].Technology of Water Treatment, 2007, 33(8): 85-87.
[9] 万亚珍. 混凝剂辅助化学沉淀法处理高含磷废水的研究[J].磷肥与复肥, 2003, 18(4): 16-17.WAN Ya-zhen, Experiments on purification of high P content waste water by chemical precipitation method associated with coagulating agent[J]. Phosphate & Compound Fertilizer, 2003,18(4): 16-17.
[10] Diefried D, Manfred S. Elimination of phosphorus from municipal and industrial wastewater[J]. Water Science and Technology, 1999, 40(4/5): 195-202.
[11] Berg U, Donnert D, Ehbrecht A, et a1. “Active filtration” for the elimination and recovery of phosphorus from wastewater[J].Colloids Surf A: Physicochem Eng Aspects, 2005, 265(1/2/3):141-148.
[12] Kostantinos K, Maximos P, Georgios N, et al. Removal of phosphate species from solution by adsorption onto calcite used as natural adsorbent[J]. Journal of Hazardous Materials, 2007,139(3): 447-452.
[13] Song Y H, Weilder P G, Berg U, et a1. Calcite-seeded crystallization of calcium phosphate for phosphorus recovery[J].Chemosphere, 2005, 63(2): 236-243.
[14] 许虹, 张静, 高一鸣. 利用矿物方解石进行水体除磷实验研究[J]. 地质前缘, 2008, 15(4): 138-141.XU Hong, ZHANG Jing, GAO Yi-ming. Experimental studies on removal of phosphor in eutrophic water bodies by utilization of mineral calcite[J]. Earth Science Frontiers, 2008, 15(4):138-141.
[15] 林建伟, 朱志良, 赵建夫, 等. 方解石去除水中磷酸盐的影响因素研究[J]. 中国给水排水, 2006, 22(15): 67-70.LIN Jian-wei, ZHU Zhi-liang, ZHAO Jian-fu, et al. Influencing factors of efficiency of phosphate removal by calcite[J]. China Water and Wastewater, 2006, 22(15): 67-70.
[16] 彭书传, 黄川徽, 陈天虎, 等. 苏皖沉积型坡缕石酸溶动力学研究[J]. 硅酸盐学报, 2004(11): 1399-1404.PENG Shu-chuan, HUANG Chuan-hui, CHEN Tian-hu, et al.Kinetics of acid-dissolution of paligorskite deposited in Jiangsu and Anhui province[J]. Journal of Chinese Ceramic Society,2004(11): 1399-1404.
[17] 张兴法. 液-固相反应薄片颗粒的宏观动力学[J]. 阜阳师范学院学报: 自然科学版, 1995(1): 6-9.ZHANG Xing-fa. The overall kinetics of the plane particle in liquid-solid reaction[J]. Journal of Fuyang Teachers College:Natural Science, 1995(1): 6-9.
