HPLC测定野老鹳草中没食子酸和鞣花酸的总量
2010-05-26吴秋月王琪琳刘丽芳
金 欣, 王 锋, 姚 岚, 吴秋月, 王琪琳, 刘丽芳
(中国药科大学中药分析教研室,江苏南京 211198)
HPLC测定野老鹳草中没食子酸和鞣花酸的总量
金 欣, 王 锋, 姚 岚, 吴秋月, 王琪琳, 刘丽芳*
(中国药科大学中药分析教研室,江苏南京 211198)
野老鹳草;HPLC;没食子酸;鞣花酸;含量测定
目的:建立野老鹳草中没食子酸和鞣花酸总量测定的方法。方法:采用HPLC法。色谱条件:色谱柱为Diamonsil C18(2)色谱柱(150 mm×4.6 mm,5 μm);流动相为乙腈-0.1%磷酸溶液梯度洗脱;流速为1.0 mL/min;检测波长为274 nm;柱温为25℃。结果:没食子酸和鞣花酸的线性范围分别是:0.075~5.00 μg(r=0.999 5)、0.05~2.00 μg(r=0.999 5),平均回收率分别是97.9%(RSD=0.53%)、102.6%(RSD=2.14%)。结论:本法简便、准确、重复性好,可用于野老鹳草药材的质量评价。
老鹳草是我国常用中药,《中国药典》2005版中收载正品老鹳草为牻牛儿苗科植物牻牛儿苗Erodium stephanianum Willd.、老鹳草 Geranium wilfordii Maxim.或野老鹳草Geranium carolinianum L.的干燥地上部分[1]。据文献报道[2]老鹳草的主要成分为鞣质、黄酮类、有机酸及挥发油等,其具有广谱抗菌、抗病毒及抗氧化等多种药理作用[3,4]。其中鞣质类化合物老鹳草素及其分解产物是抗氧化作用的主要成分,具有抗氧化、维持体内自由基的稳定和平衡、消除有害的自由基反应、中断脂质过氧化、减少脂质过氧化产物等作用[5,6]。没食子酸和鞣花酸为老鹳草素的主要分解产物,本文采用高效液相色谱法同时对目前我国老鹳草药材的主流品种——野老鹳草中没食子酸和鞣花酸的总量进行了测定。该研究为制定野老鹳草的质量标准和对野老鹳草药材的进一步开发利用提供了科学依据。
1 仪器与试药
Agilent 1100 HPLC Series液相色谱仪(Agilent公司,美国);Diamonsil C18(2)色谱柱,(150 mm×4.6 mm,5 μm)(Dikma 公司,美国);KH3200E 型超声波清洗器(昆山禾创超声仪器有限公司);没食子酸对照品(中国药品与生物制品检定所,批号:110831-200302);鞣花酸对照品(自制,纯度大于98%(HPLC))。老鹳草全部由中国药科大学中药学院刘丽芳副教授鉴定为野老鹳草Geranium carolinianum L.,标本存放于中国药科大学中药分析实验室;乙腈为色谱纯(Dikma公司,美国);水为乐百氏纯净水;其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件 色谱柱采用Diamonsil C18(2)色谱柱,(150 mm ×4.6 mm,5 μm)。流动相为乙腈-0.1%磷酸溶液,洗脱程序见表1。流速1.0 mL/min,检测波长274 nm,柱温25℃。色谱图见图1。
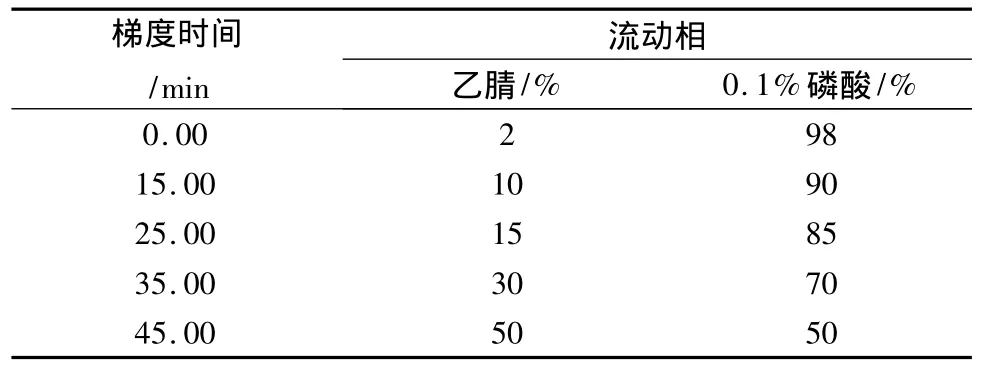
表1 洗脱程序
2.2 溶液的制备

图1 没食子酸(A)、鞣花酸(B)、野老鹳草(C)的HPLC色谱图
2.2.1 供试品溶液的制备 取本品细粉约1 g(过4号筛),精密称定,置锥形瓶中,加盐酸-50%甲醇(10%(v/v))50 mL,加热回流0.5 h,滤过。残渣同法再提取1次,滤过,合并滤液,减压蒸干溶剂,残渣用50%甲醇溶解并定容至50 mL量瓶中,摇匀,精密移取10 mL蒸干,残渣用50%甲醇溶解并定容至25 mL量瓶中,摇匀,用微孔滤膜(0.45 μm)滤过,取续滤液,即得。
2.2.2 对照品溶液的制备 精密称取没食子酸对照品适量,加50%甲醇制成每1 mL中含50 μg的溶液;精密称取鞣花酸对照品适量,加二甲基亚砜制成每1 mL中含50 μg的溶液,即得。
2.3 标准曲线的绘制
2.3.1 没食子酸标准曲线的绘制 精密吸取0.25 mg/mL的没食子酸对照品溶液0.15、1.00、3.00、5.00、7.00 mL加50%甲醇稀释定容至10 mL,摇匀,分别进样20 μL,用上述色谱条件测定。以进样量(μg)为横坐标,测得的峰面积积分值为纵坐标绘制标准曲线,回归方程:Y=2 752.5X+54.156,r=0.999 5。结果表明在0.075~5.00 μg之间有良好的线性关系。
2.3.2 鞣花酸标准曲线的绘制 精密吸取0.10 mg/mL的鞣花酸对照品溶液 0.25、1.50、2.50、3.50、5.00 mL加二甲基亚砜稀释定容至10 mL,摇匀,分别进样20 μL,用上述色谱条件测定。以进样量(μg)为横坐标,测得的峰面积积分值为纵坐标绘制标准曲线,回归方程:Y=2 998.7X+44.347,r=0.999 5。结果表明在0.05~2.00 μg之间有良好的线性关系。
2.4 稳定性试验 精密吸取供试品溶液(南京(080604)),分别于 0、3、6、9、12、24 h 测定,没食子酸RSD=2.48%,鞣花酸RSD=4.65%,表明供试品溶液在24 h内稳定。
2.5 精密度试验 精密吸取等体积的对照品溶液注入液相色谱仪,重复6次,没食子酸RSD=0.34%,鞣花酸RSD=2.23%,表明仪器精密度良好。
2.6 重复性试验 取同一样品(南京(080508))5份,按“2.2.1”项方法制备,在2.1项条件下测定,总没食子酸含量分别为0.305 9%、0.311 8%、0.317 5%、0.310 5%、0.305 0%,平 均 值 为0.310 1%,RSD=1.62%;总鞣花酸含量分别为0.106 5%、0.103 4%、0.102 6%、0.106 8%、0.105 1%,平均值为0.104 9%,RSD=1.77%,表明方法的重复性良好。
2.7 检测限及定量限 分别将没食子酸和鞣花酸对照品溶液逐渐稀释后进样,取S/N=3时的浓度为检测限,经测定,没食子酸检测限为0.075 μg/mL,鞣花酸检测限为0.25 μg/mL;取S/N=10时的浓度为定量限,经测定,没食子酸定量限为0.25 μg/mL,鞣花酸定量限为 0.70 μg/mL。
2.8 回收率试验 精密称取已知含量的样品(山东(0708200308))共6份,分别精密加入一定量的没食子酸对照品和鞣花酸对照品,混匀后,按2.9项方法测定含量,计算平均回收率,结果见表2。

表2 回收率测定结果(山东(0708200308))
结果表明,本法具有较好的准确性。
2.9 样品的含量测定 取14批样品,经干燥、粉碎后,按2.2.1项方法处理。精密吸取供试品溶液,进样20 μL,用外标法测定含量,结果见表3。
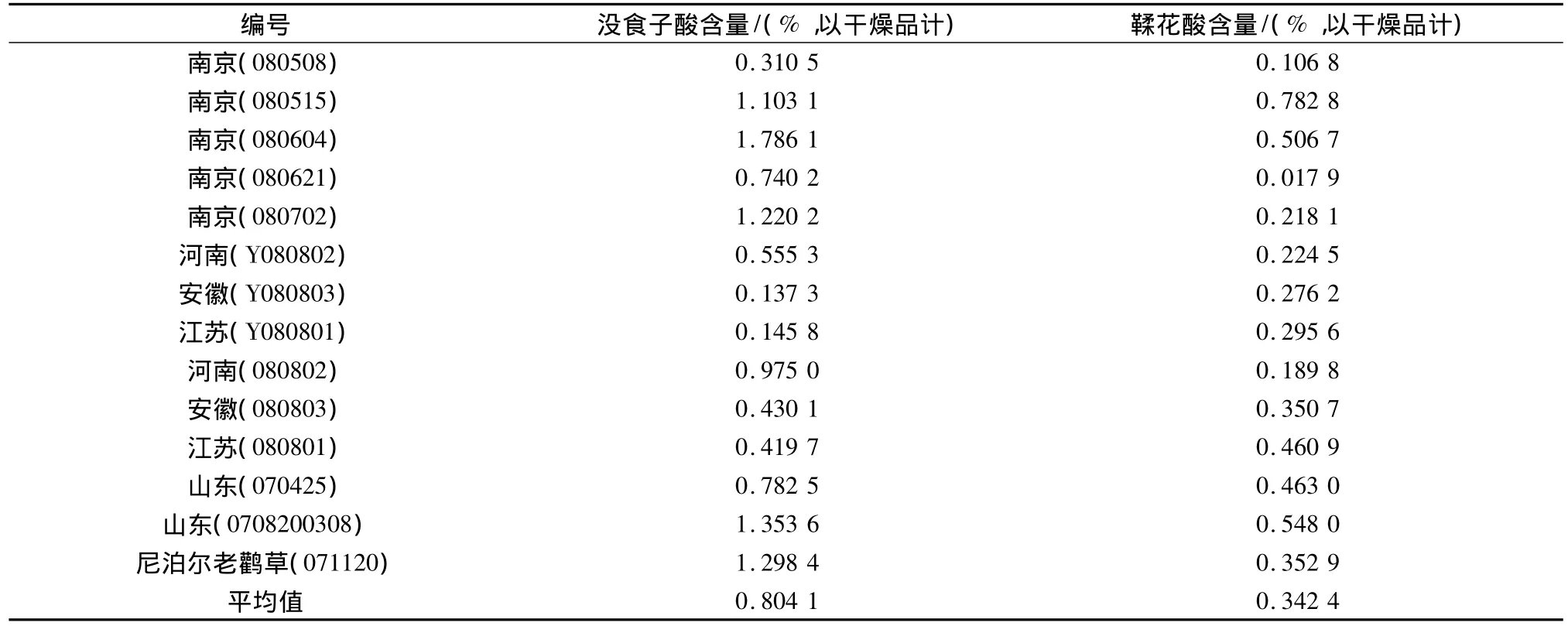
表3 样品测定结果
3 讨论
3.1 精密称取同一批样品9份,分别考察了时间为0.5、1、2 h,盐酸浓度为 5%、10%、20%,溶剂为30%甲醇、50% 甲醇、70% 甲醇,体积为 25、50、75 mL的4因素3水平的正交试验,结果表明,时间为0.5 h,盐酸浓度为10%,溶剂为50%甲醇,体积为50 mL,提取2次为最优方案。
3.2 老鹳草素(geraniin)属于可水解鞣质中的鞣花鞣质(ellagitannin),加水分解时分解为没食子酸(gallic acid)、六羟基联苯二甲酸(hexahydroxydiphenic acid)、鞣花酸(ellagic acid)、柯里拉京(corilagin)、云实酸(brevifolincarboxylic acid)、云实素(brevifolin)[5],见图 2。

图2 老鹳草素及其分解产物
据文献报道[6-8],没食子酸对结肠运动呈现显著的抑制作用,因此老鹳草总鞣质(HGH)有较好的治疗腹泻作用;鞣花酸具有抗氧化功能,抗癌、抗突变性能,对人体免疫缺陷病毒有抑制作用。本次实验采用高效液相色谱法同时对野老鹳草中没食子酸和鞣花酸的含量进行测定。我们在实验中发现,野老鹳草的50%甲醇提取物中成分组成非常复杂,样品不经水解直接进样分析,70 min内有包括上述两种待测成分在内的80多个色谱峰被分离出来,因此,样品液不经水解直接分析所需时间长,且对色谱柱的柱效要求非常高,给分析测定带来一定的难度。两种含量相对较高的待测成分平均值也仅为0.125 2%和0.175 8%。而将样品水解后,由于多种多酚类化合物具有相同的水解产物,因而使样品溶液所含成分大大减少,所需分析时间缩短,柱效要求更适中,水解后样品中没食子酸和鞣花酸总量平均值达到0.804 1%和0.342 4%。综合上述原因,我们将该药材水解后再进行含量测定。
3.3 我们从各地共收集药材样品14批,除一批从云南药材公司购置的鲜药材经鉴定为尼泊尔老鹳草(G.nepalense Sweey.)外,其它药材均为野老鹳草(G.carolinianum L.)的干燥地上部分。由此说明,该种目前为老鹳草药材的主流品种。样品测定结果表明,各地野老鹳草水解后的没食子酸含量在0.137 3% ~1.786 1%之间,平均值为0.804 1%;鞣花酸含量在0.017 9% ~0.782 8%之间,平均值为0.342 4%。各批药材中没食子酸和鞣花酸含量的差异可能与产地气候,采收期等因素有关,值得进一步探讨。
[1]中国药典[S].一部.2005:80.
[2]金晴昊,崔京浩,郭建鹏.老鹳草的研究进展[J].时珍国医国药,2005,16(9):840-841.
[3]Küpeli E,Tatli I I,Akdemir Z S,et al.Estimation of antinociceptive and anti-inflammatory activity on Geranium pratense subsp.Finitimum and its phenolic compounds[J].J Ethnopharmacol,2007(114):234-240.
[4]LI Jiyang,Huang Hai,Zhou Wei,et al.Anti-hepatitis B Virus Activities of Geranium carolinianum L.Extracts and Identification of the Active Components[J].Biol Pharm Bull,2008,31(4):743-747.
[5]杜晓鸣,郭永泗.老鹳草素及抗氧化作用[J].国外医药·植物药分册,1990,5(2):57-61.
[6]邹建华.尼泊尔老鹳煎液成分对离体肠管运动的影响[J].国外医学·中医中药分册,1993,15(4):44-45.
[7]王丽敏,卢春风,路雅真.老鹳草鞣质类化合物的抗腹泻作用研究[J].黑龙江医药科学,2003,26(5):29-30.
[8]丁运生,孙小虎,李有桂,等.鞣花酸及其衍生物研究进展[J].合肥工业大学学报(自然科学版),2008,31(11):1809-1812.
Determination of gallic acid and ellagic acid in Geranium carolinianum L.by HPLC
JIN Xin, WANG Feng, YAO Lan, WU Qiu-yue, WANG Qi-lin, LIU Li-fang*
(Department of Analysis of Chinese Medicine,China Pharmaceutical University,Nanjing 211198,China)
Geranium carolinianum L.;HPLC;gallic acid;ellagic acid;determination
AIM:To establish a method for determining gallic acid and ellagic acid in Geranium carolinianum L..METHODS:An HPLC method was performed on a Diamonsil C18column(150 mm ×4.6 mm,5 μm)with acetonitrile-0.1%H3PO4as mobile phase.The flow rate was 1.0 mL/min,the detection wavelength was set at 274 nm and the column temperature was at 25℃.RESULTS:The calibration curve of gallic acid and ellagic acid were linear in the range of 0.075 ~5.00 μg(r=0.999 5)and 0.05 ~2.00 μg(r=0.999 5),respectively.The average recovery of gallic acid and ellagic acid were 97.9%(RSD=0.53%)and 102.6%(RSD=2.14%),respectively.CONCLUSION:The method is simple,accurate and suitable for the quality control for this herb.
R284.1
A
1001-1528(2010)07-1172-05
2009-09-18
国家药典委员会2010版一部标准研究课题(YS-229,YS-230)
金 欣(1985-),女,在读硕士研究生,主要从事中药制剂分析研究。Tel:(025)86185373
*通讯作者:刘丽芳(1969-),女,博士,副教授,主要从事中药活性成分和动物药研究。Tel:(025)86185446 E-mail:liulifang69@126.com
