如何实施对检测实验室检验能力的确认
2010-05-03倪敏喻雨琴
文/倪敏 喻雨琴
实验室是用于在科学上为阐明某一现象而创造特定的条件以便观察其变化和结果的机构。检测实验室则是用于完成检测工作的实验室,向社会提供准确可靠的检测数据和结果,它在技术经济活动和社会发展过程中都占有重要的地位。检测实验室终产品的体现是检测报告,实验室要向社会出具高质量的报告,并得到社会各界的信赖和认可,所以其检测数据的可靠性至关重要。
实验室是否通过检查并提供客观证据,证实某一特定预期用途的特定要求得到满足?实验室是否对非标准方法、实验室设计(制定)的方法、超出其预定范围使用的标准方法、扩充和修改过的标准方法进行确认,以证实该方法适合于预期的用途?CNAS-CL01《检测和校准实验室能力认可准则》提出了方法的确认。
确认的定义:通过检查并提供客观证据以证实某一特定的预期用途的特殊要求或应用要求已得到满足的认定。确认应尽可能的广泛全面,以满足预定用途或应用领域的需要,证实实验室能够正确地执行标准方法。实验室是否记录所获得的结果、使用的确认程序以及该方法是否适于预期用途的声明?这就要求检测实验室在开始检测之前应确认能够正确地运用标准方法;当标准的方法发生了变化,应重新进行确认,以证实实验室能够正确地执行变化了的标准方法。
一般实验室应对4种检测方法进行确认,以证实这些方法适用于预期用途,包括:标准方法应用实施前;非标准方法;自己新设计开发;超过预期范围使用的标准方法/扩充和修改过的标准方法。方法确认的前提是验证,验证则是一个持续的过程。
确认是运行前和变化后实施的评定,目的在于证明各检测/验方法能力能够达到预期的控制水平(或满足可接受水平);验证是在运行中和运行后进行的评定,目的在于证明确实达到了预期的控制水平(和/或满足了可接受水平)。
一、检测方法确认的技术要求说明
目前对检测方法在使用前或变更后的确认可依据GB/T 27404-2008《实验室质量控制规范食品理化检测》附录F对技术要素进行验证。
1.回收率
回收率是向试样中加入已知量被测组分的标准物质,最后看加入已知量的标准物质是否能定量回收,以判断分析过程是否存在系统误差。在样品中加入标准物质,测定其回收率,可以检验分析方法的准确程度和样品所引起的干扰误差。
对于食品中的禁用物质,回收率应在方法测定低限、2倍方法测定低限和10倍方法测定低限进行三水平试验;对于已制定最高残留限量(MRL)的,回收率应在方法测定低限、MRL、选一合适点进行三水平试验;对于未制定MRL的,回收率应在方法测定低限、常见限量指标、选一合适点进行三水平试验。回收率的参考范围见表1:
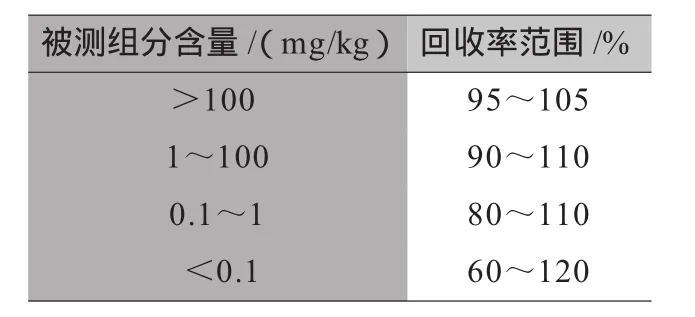
表1 回收率范围
回收率公式:
回收率(%)=(添加标准物质的试样实测量-试样中被测物量)/加入标准物质量×100%---(1)
2.校准曲线
①被测组分浓度的选择
应描述校准曲线的数学方程以及校准曲线的工作范围,浓度范围尽可能覆盖一个数量级(如0.1~1.0),至少作5个点(不包括空白),在不同浓度水平上画出一条标准曲线。对于筛选方法,线性回归方程的相关系数不应低于0.98;对于确证方法,相关系数不应低于0.99。测试溶液中被测组分浓度应该在校准曲线的线性范围内。
对于给出良好线性度方法,用测量标准在5个不同浓度水平上画出一条标准曲线即可。当线性度差时,需要更多个测量标准。定量分析方法的线性都是通过分析具有覆盖方法所要求浓度范围的分析物样品来确定,应用最小二乘法将其结果用于计算回归线。如果某一检测方法在一特定范围内是线性的(但不是绝对要求),这就比较理想。
标准曲线回归方程:
y=a+bx -------------------------(2)
相关系数R=0.9999
式(2)中:a——截距;
b——斜率。
在分析测试中常会遇到处理两个变量之间的关系,如在建立校准曲线时需要了解被测组分的浓度和响应值之间的关系。两个变量x与y之间的关系存在着函数关系,y值随着x值按照确定的规律变化,由一个xi值可以精确地求出yi值。
(x,y)的散布图 y=f(x)------------(3)
回归函数是一个线性函数,则称变量间是线性相关的。一元线性回归方程:y=a+bx
由于存在测定误差,x与y不是函数关系,而是相关关系(相关关系是统计关系)。
②校准曲线的绘制注意事项
每种应用校准曲线的分析方法在初次使用时,可通过绘制校准曲线以确定它的检测上限,并结合检测下限确定其检测范围。绘制校准曲线应注意:
a校准曲线一般可根据4~6个浓度及其测量信号响应值绘制;
b测量信号值的最小分度应与纵坐标的最小分格相适应;
c浓度的整数值应落在横坐标的中格或大格的粗线上,以便于分小格查阅,并尽量使校准曲线的几何斜率接近于1(与横坐标约成45°角),以使在两个轴上的读数误差相近。
d校准曲线的斜率常因温度、试剂批号等条件的变化而变化。在测定未知样品的同时测绘校准曲线是最理想的。否则,应在测定未知样品的同时,平行测定线性范围内的中等浓度标准溶液和空白溶液各2份,取均值相减后,与以前绘制的校准曲线上相同点进行核对,二者的相对差值根据方法精密度要求小于5%或小于10%,否则应重新绘制校准曲线。
③校准曲线的相关因素
绘制校准曲线所依据两个变量的线性关系,决定着校准曲线的质量和样品测定结果的准确度。影响校准曲线线性关系的因素:
a分析方法本身的精密度;
b分析仪器的精密度;
c量取标准溶液所用量器的准确度;
d易挥发溶剂(如萃取反应显色物的有机溶剂)挥发所造成比色液体体积的变动幅度;
e分析人员的操作水平。
由于存在测定误差,可用“相关系数”来判断校准曲线的线性关系。如用4~6个浓度的标准溶液及其测量响应值绘制的校准曲线,根据实验经验应力求其相关系数的绝对值IrI≥0.999。否则可参照上述影响线性关系的诸因素,找出原因并尽可能加以纠正,重新测定和绘制新的校准曲线。
对于成熟的分析方法和熟练的分析人员若能细心并严格按规范操作,则校准曲线的相关系数绝对值IrI≥0.999,在实验条件不变情况下校准曲线的斜率一般很稳定,其截距等于零。
④相关系数检验法检验
方程y=a+bx中因变量y与自变量x之间是否确实存在相关关系,在求线性回归方程的过程中也可用最小二乘法求得“回归方程”,画出一条直线,这可用相关系数检验法检验。相关系数的显著检验具体步骤如下:
a按照公式(4)由样本值计算相关系数r
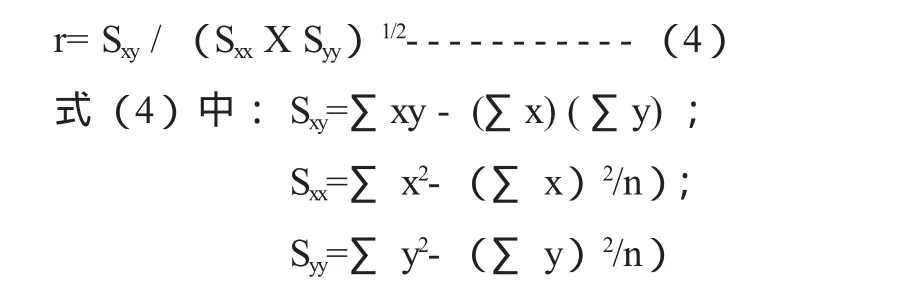
b给定显著性水平α,按照样本容量n,由“相关系数临界值表”(见“回归分析”中的“相关系数临界值表”)中查出临界值rα,n。
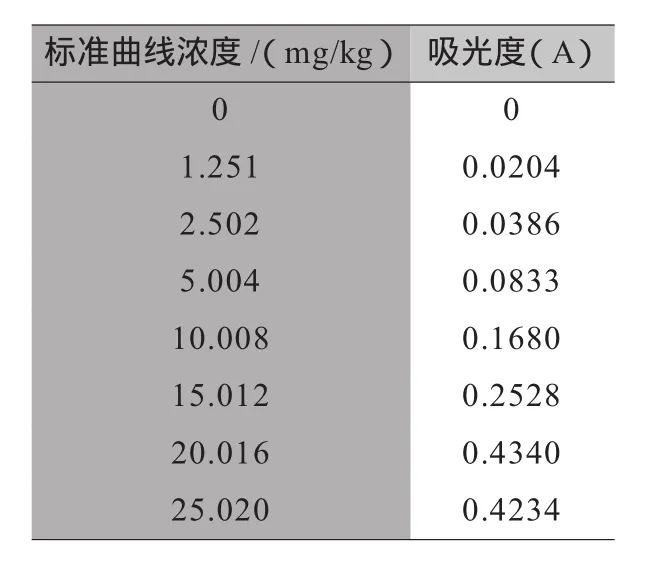
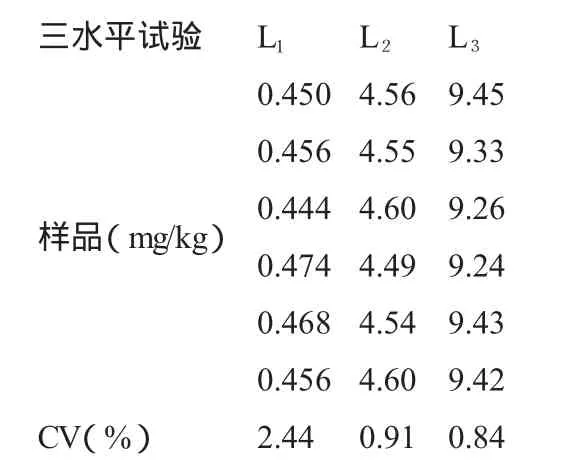
c 比较IrI与rα,n的大小,若IrI≥rα,n,则认为x与y之间存在线性相关关系;若IrI 例:用原子吸收分光光度法测定微量钴,得到下列一组数据(见表2): 表2 用原子吸收分光光度法测定微量钴 试由上述数据确定A与c的线性回归方程式,该关系式是否有意义? 解:分别求出斜率b和截距a 按下式计算出相关系数:r=Sxy/(SxxXSyy)1/2=1.000 查相关系数临界值表,r0.05,5=0.878,r>r0.05,5,说明确立的回归方程是有意义的。 精密度就是几次平行测定(分析)结果相互接近的程度。精密度的高低用偏差来衡量。其中偏差是指单次测定结果值与几次测定结果平均值之间的差别。 ①偏差的计算 对于食品中的禁用物质,精密度实验应在方法测定低限、2倍方法测定低限和10倍方法测定低限三个水平进行; 对于已制定MRL(最高残留限量)的,精密度实验应在方法测定低限、MRL、选一合适点三个水平进行;对于未制定MRL的,精密度实验应在方法测定低限、常见限量指标、选一合适点三个水平进行。 复测定次数至少为6。实验室内部的变异系数参考范围见表3。 表3 实验室内变异系数 绝对偏差=单次测得值-测得结果平均值 相对偏差=绝对偏差/测得平均值×100% 平均偏差:各次测量值偏差的绝对值之和被测量次数除的平均值。 相对平均偏差:平均偏差/平均结果x 100% 极差:也称全距R,是一组数据极大值与极小值之差。 目前普遍采用标准差的相对值来反映数据的相对波动度,标准差的相对值称为相对标准偏差RD。 单次测量结果的相对标准偏差又称变异系数CV。 例:测定试样中的氟化物含量,重复测定5次,其数据为: 0.45;0.39;0.47;0.40;0.43 mg/L,试求极差?平均偏差?标准差?变异系数? 计算结果:极差R=0.47-0.39=0.08 mg/L 平均偏差d=I0.13I/5=0.026 mg/L 标准差S=0.00451/2/(5-1)=0.033 mg/L 变异系数CV=0.033/0.43x100%=7.7% ②标准差的作用 a表示观察值分布的离散程度(由偶然误差决定)。当两组观察值在单位相同、两均数相近的条件下,标准差较大,说明观察值的变异程度较大,即观察值围绕均数的分布较离散,均数的代表性较差;反之均数的代表性就较好。 在理化检验工作中,例如摸索某一检验方法的实验条件或比较两种检测方法时,都常用到标准差。 b用标准差计算变异系数。当两组观察值单位不同,或两均数相差较大时,不能直接用标准差比较其变异程度的大小,这时可用变异系数(CV)进行比较。 同标准差一样,变异系数愈小,说明观察值的变异程度愈小;变异系数愈大,说明观察值的变异程度愈大。理化检验方法的精密度就是用变异系数28 ssm /2010/5(或称相对标准差)表示的。 重复性试验:目的是为了预测精密度。 平行测定的次数愈多,其平均值愈接近真实值。合理的反映实验室对一个分析方法的精密度应该通过在一个时间阶段(1~2周)内若干批的试验中得到,这样可以区分出批内和批间的变异而指出产生随机误差的重要来源。另一方面从所有批的批内标准差合并值可以指出常规分析中随机误差的规律。 在实验设计中必须选择合适的批数m和重复测量次数n,一般情况下n=2~4次,m=5~10次为最合适。 ①测定方法 方法的测定低限按公式(9)计算: 式中:CL-方法的测定低限; Sb-空白值标准偏差(一般平行测定20次得到); b-方法校准曲线的斜率。 对于已制定MRL(最高残留限量)的物质,方法测定低限加上样品在MRL处标准偏差的3倍,不应超过MRL值。对于禁用物质,方法测定低限应尽可能低。 测定低限又称检出限或检测限:把3倍空白值的标准偏差(测定次数n≥20)相对应的质量或浓度称为测定低限(检出限)。如色谱法:设色谱仪最低响应值和检出限分别按公式(10)和(11)计算: 式中:N为仪器噪音水平。 式中:b-标准曲线回归方程中的斜率; 响应值 /μg或响应值 /ng; S-仪器噪音的3倍,即仪器能辨认的最小物质信号。 ②测定低限(又称检出限或检测下限)分类 测定低限可分为三类: a仪器检测下限:即相当于背景,仪器检测的可靠最小信号,通常用信号噪声比表示,当N/S≥3时,定义为仪器检测下限; b方法检测下限:即某方法可检测的最小浓度。 方法的测定低限(检测下限)按公式(9)计算;标准偏差按公式(12)计算: 标准曲线回归方程按公式(2)计算。 c样品检测下限:即相对于空白可检测的最小样品的含量。定义样品检测下限为3倍空白标准偏差即3S空白,故而只有空白含量为零时,样品检测下限才等于方法检测下限,然而空白含量往往不等于零。 准确度——表示分析结果与真实值接近的程度。 重复分析标准物质(实物标样)或水平测试样品,测定含量(经回收率校准后)平均值与真实值的偏差指导范围(见表3)。 表3 测定值与真实值的偏差指导范围 分析结果和真实值之间的差值称为误差,误差越小分析结果的准确度越高;误差越大分析结果的准确度越低。 准确度的高低用误差来衡量。 误差分为: 绝对误差——测定值与真实值之间的差值; 相对误差——测定值与真实值之间的差值对真实值的百分数。 例如:某物质的真实值为1.0000g,测定值为0.9980g, 其绝对误差为-0.0020g; 其相对误差=(-0.0020/1.0000)×100%=-0.2%。 将测得的组分的百分含量与公认的“真实值”之差,作为定量分析的误差,以衡量某一分析方法或某一实验室的分析水平,或衡量某一分析者的技术熟练程度。 相对误差是误差在真实值中所占的百分率: 相对误差=(绝对误差/被测量真实值)×100%。 提取效率可用以下方法进行试验: ① 用阳性的标准物质或水平测试的阳性样品进行试验; ②阳性样品用同一溶剂反复提取,观察被分析物的浓度变化; ③用不同提取技术或不同提取溶剂进行比较。 提取效率与样品的前处理、选择的溶剂、提取技术有关:由于食品本身(如蛋白质、脂肪、糖类等)对分析测定常产生干扰,因此在定性定量分析测定之前必须对样品进行前处理。样品在处理过程中,既要排除干扰的因素,又不致于使被测物受到损失,而且还应能使被测物达到一定浓度,从而使测定得到理想结果。所以在分析过程中,样品的前处理、选择的溶剂、提取技术是分析测定的重要步骤。在分析化学中,常用的分离方法有沉淀分离法、液-液萃取分离法、离子交换分离法、色谱分离法、蒸馏和挥发分离法等。 如液-液萃取分离又叫溶剂萃取分离,一般简称萃取分离。这种方法是利用与水不相混的有机溶剂同试液一起震荡,这时,一些组分进入有机相中,另一些组分仍留在水相中,从而达到分离的目的。提取效率直接影响到检测回收率、精密度、准确度、线性范围和最低检出限。 对于检测筛选方法和确证方法特异性必应予以规定,尤其对于确证方法必应尽可能清楚地提供待测物的化学结构信息,特别是一些化学官能团、分子结构仅基于色谱分析而没有使用分子光谱测定的方法,不能用于确证方法。确证方法可采用: ①气相色谱-质谱; ②液相色谱-质谱; ③免疫亲和色谱或气相色谱-质谱; ④气相色谱-红外光谱; ⑤液相色谱-免疫层析。 笔者曾见一检测实验室在验证确认其新研究的检测方法时仅用分光光度法,当介绍到分光光度计在两个不同波长处分别出现有两个峰时,实验人员以“峰形漂亮对称”为理由认为这就是要确定选择的具特异性化合物的峰,但该实验室并未采用其他的方法作佐证进行定性,这是没有科学依据的确证方法。因此策划采用不同的仪器作为确证方法,可以清楚地提供待测物的化学结构信息,特别是一些化学官能团、分子结构,可确保有效地进行定性。 检测方法应具有对可变试验因素的抗干扰能力,当测定条件发生细小变动时,方法应具有一定的保持测定结果不受影响的承受程度。 耐用性也称抗变性或稳健性。不同实验室在使用同一检测方法时,他们不可避免地会在程序方法上引入一些小的变动,这些变动可能会也可能不会对方法的工作特点产生重大影响。一种方法的耐用性(抗变性或稳健性)是通过慎重地对检测方法引入某些小的改变并检查其影响结果来测试的。用同一样品由三个以上实验室按同样检测方法检验,该检测方法应具有一定的保持测定结果不受影响的承受程度。 再现性(复现性)—表示不同分析人员或不同实验室之间在各自实验条件下所得分析结果的精密度。一个数值R在再现性条件下,两次测试结果的绝对差值不超过此数R的概率为一个指定值(一般为95%)。所谓再现性条件是指在两个不同实验室,不同操作人员,不同仪器设备,在不同或相同的时间内,用同样试验方法程序对同一被试样品所得的两个独立单次试验结果的再现试验条件。也可理解为在改变了的测量条件下,同一被测量结果之间的一致性。 确认研究新的检测方法,其研究必须要提供有按7特异性和8耐用性两个技术要素指标的数据。综合了上述8个技术参数验证数据才能确认该检测方法预期目的有效性。 如:首次采用的标准检测方法在应用于样品检测前,应对检测方法的技术要素进行验证确认。 首次采用的标准方法在应用于样品检测前,应对检测方法的技术要素进行验证试验,再实施确认。如某检测实验室要扩SC/T 3025-2006《水产品中甲醛的测定》检测方法,该方法适用范围:适用于水产品中甲醛含量的定量测定。 ①立项申请:目的、人员、仪器设备及试剂、时间安排及进度; ②计划任务书:人员、时间安排及进度、策划验证项目、判定值、评价、确认的方法; ③验证工作的实施; ④评价和确认。 ①回收率 “对于食品中的禁用物质,回收率应在方法测定低限、两倍方法测定低限和十倍方法测定低限进行三水平试验。” 根据NY 5172-2002《无公害食品水发水产品》规定,甲醛为食品中的禁用物质,其最高残留限量(MRL)≤10.0mg/kg。 由此三个添加回收率水平是 a.方法测定低限0.5mg/kg; b.MRL 10.0mg/kg; c.MRL与方法测定低限间合适点5.00mg/kg。 按SC/T 3025-2006《水产品中甲醛的测定》检测方法4.1.5.1对甲醛标准储备液的标定,甲醛标准溶液的浓度是5.004mg/L; 计算得:a XL1-1=0.450mg/kg;XL1-2=0.456mg/kg; 按照标准GB 27404-2008附录F.1回收率规定:被测组分含量在0.1mg/kg~1mg/kg,其回收率范围80%~110%;1.0mg/kg~100mg/kg其回收率范围90%~110%。故本项目回收率符合标准要求。 ②校准曲线 曲线Y=0.0171X-0.0018 R=0.9999 按照标准GB 27404-2008附录F.2校正曲线要求,相关系数R>0.98,故本项目能满足标准的要求。 ③精密度 同回收率部分进行三水平试验,每个水平重复6次,对变异系数进行考查。 三水平试验 L1 L2 L3 0.450 4.56 9.450.456 4.55 9.330.444 4.60 9.260.474 4.49 9.240.468 4.54 9.430.456 4.60 9.42 CV(%) 2.44 0.91 0.84样品(mg/kg) 按GB 27404-2008附录F.3精密度的要求: 被测组分含量范围在 1mg/kg 10mg/kg 10mg/kg 变异系数应 <11%<7.5% <7.5% 故本项目能满足标准的要求。 ④测定低限 方法的测定低限按公式(9)计算: CL=3Sb/b 对于已制定MRL的物质,方法测定低限加上样品在MRL处,标准偏差的3倍,不应超过MRL值。对于禁用物质,方法测定低限应尽可能低。 平均测定20次空白,其吸光度为:0.0010,0.0012,0.0012,0.0008,0.0009,0.0011,0.0010,0.0012,0.0008, 0.0012, 0.0014, 0.0008, 0.0011, 0.0013,0.0012,0.0010,0.0009,0.0007,0.0010,0.0009 标准偏差(n=20) Sb=0.000190,标准曲线斜率=0.0171 CL=3Sb/b=3×0.000190/0.0171=0.0333mg/kg 当取样量为10g,稀释倍数为10,标样与样品比色体积都为10mL时,CL=0.0333×10=0.333mg/kg, 低于SC/T 3025-2006《水产品中甲醛的测定》中(4.1)检出限0.50mg/kg的要求,故能满足最低检出限的要求。 ⑤准确度 取阴性样品添加甲醛标准溶液,添加量为10.008mg/kg; 取平均样测定: m1=10.00g m2=10.00g 甲醛标准溶液浓度为 5.004mg/L A1=0.0151、A2=0.0152 测定值与真实值之间的差值对真实值的百分数 (|9.91-10.008|/10.008)×100%=0.98% <15% 按照标准GB 27404-2008附录F.5准确度的要求,故本项目能满足标准的要求。 ⑥提取效率 取甲醛标准溶液按照标准方法处理,收集蒸馏液,取10.0mL进行比色反应,上仪器读吸光度;另取甲醛标准溶液2.00mL,稀释至10.0mL进行比色反应,上仪器读吸光度,得:未经蒸馏提取甲醛标准溶液的吸光度: A 0.1688 X 9.93mg/kg 经蒸馏提取甲醛标准溶液的吸光度: A 0.0151 X 9.88mg/kg 经蒸馏提取后的残液,再次蒸馏提取甲醛测得吸光度: A 0.0003 X 1.2mg/kg 在蒸馏提取处理过程中第一次收集了约99.5%,约0.5%的甲醛在第二次蒸馏提取处理中收集到。该方法提取效率本实验能满足要求。 通过对以上技术要素的验证,实验室可以确认该检测方法能达到预期使用的效果。由实验室技术负责人批准该检测方法列入实验室的扩项申请。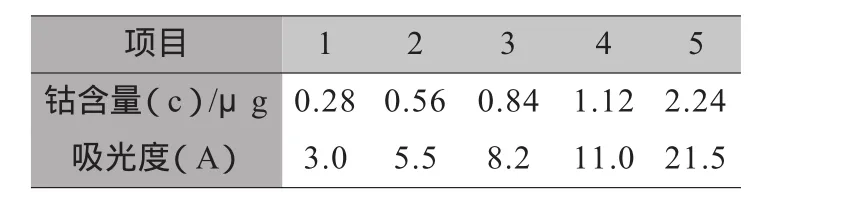

3.精密度





4.测定低限




5.准确度

6.提取效率
7.特异性(定性要求)
8.耐用性
二、实验室拟扩项标准检测方法
1.策划
2.实验人员遵循技术要素开展

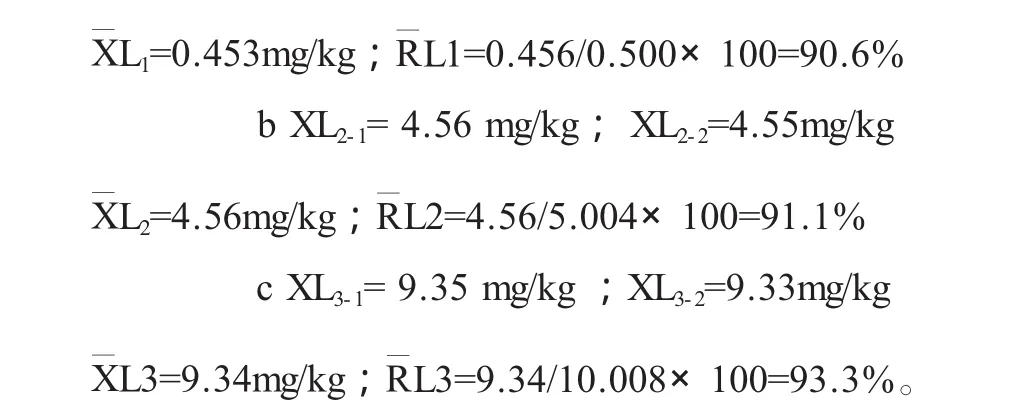

三、结论
