基于比较基因组学重构细菌的代谢途径和调控网络
2010-02-09杨琛
杨琛
上海生命科学研究院植物生理生态研究所 中国科学院合成生物学重点实验室,上海 200032
基于比较基因组学重构细菌的代谢途径和调控网络
杨琛
上海生命科学研究院植物生理生态研究所 中国科学院合成生物学重点实验室,上海 200032
微生物基因组学的迅速发展为从功能基因与蛋白、网络及其调控等不同的角度,全面理解与认识微生物的代谢过程、构建细胞工厂,提供了丰富的背景信息。基于基因组序列进行代谢网络重构,有助于发现新的代谢功能基因、调控元件、甚至新的代谢途径,从而优化设计细胞内从原料到产品的生物合成路线。然而,目前公共数据库平台中代谢途径基因的功能注释,许多是错误或不完整的。以下着重介绍了近年来出现的一些用于代谢途径和调控网络重构的新型比较基因组学技术,并以近期的丙酮丁醇梭菌中木糖代谢途径的重构工作为例来说明其应用。
比较基因组学,代谢途径重构,调控网络重构,基因组上下文分析
随着FASTA和BLAST等搜索工具的发展、及一些公共数据库平台 (如 GenBank、Swiss-Prot等)的不断完善,基于序列同源比对来推断基因功能是当前普遍使用的基因组注释方法[3]。一些模式微生物 (如大肠杆菌、枯草芽胞杆菌、酿酒酵母等) 的功能基因组学研究的不断深入发展也为此做出了重要贡献。虽然序列同源比对的方法在很多时候是成功的,但是在一些情况下 (如被非同源基因所取代时)这种方法不能推测基因的功能,或只能给出不准确甚至错误的功能注释。这种情况给基于基因组序列的代谢网络重构造成了困难。由于单纯基于序列相似性的方法存在着局限性,近年来出现了一些比较基因组学的新技术,通过尽量发掘、综合基因组中蕴藏的相关重要信息,如基因簇[4]、调节位点共享[5]等,以准确预测基因的功能,有效解决缺失基因问题 (Missing genes,即代谢元件应该存在却还没有找到对应基因)[2];在此基础上,设计实验以验证重要的基因功能预测,最终实现代谢网络的重构。这种有机结合湿实验 (Wet experiment,指通常的实验室实验) 与干实验 (Dry experiment,指生物信息学分析) 的方法,一方面通过实验验证使基因的功能分析更加准确可靠;另一方面能够大大减轻实验工作量、提高工作效率。不少研究表明,使用这种方法,能够显著提高基因功能注释和代谢网络重构的质量,并且发现新的功能基因[6-8]、甚至新的代谢途径[9-11]。
本文将对用于代谢途径和调控网络重构的若干新型比较基因组学技术进行介绍,并举例说明它们的应用。本文的主要目的之一是鼓励实验工作者利用这些技术来解决所感兴趣的代谢途径中存在的缺失基因问题,发现新的代谢功能基因和调控元件,从而加强对代谢过程的理解和应用。
1 代谢途径和调控网络重构的比较基因组学技术
1.1 代谢途径重构
基于微生物基因组的信息解析代谢网络,识别和表征底物利用与产物合成的关键功能基因和蛋白,是进行代谢途径优化和改造的基础。虽然目前一些公共数据库平台 (如KEGG http://www.genome. jp/kegg/等) 已经注释了不少代谢基因,包括结构基因和调节基因,但是由于这些数据库往往仅根据序列同源性来推断基因的功能,因此存在着许多问题。例如许多梭菌 (如丙酮丁醇梭菌等) 的木糖代谢途径在KEGG中的注释是不完整的,甚至存在着相当多的错误。造成这种情况的原因是在生物转化过程中起重要作用的反应酶 (如糖激酶) 往往存在多个同源蛋白,它们在不同微生物中可能具有不同的生理功能,服务于不同的代谢过程,因此如果仅仅基于序列同源比对,就只能给出不准确或不完整的基因功能注释。此外,已有研究表明,即使与模式菌亲缘关系相近的种类,它们的代谢途径也常常存在着不同,如不同的营养物质摄入机制[12]、反应酶被非同源蛋白所取代[7]等,在这些情况下序列对比的方法就不能推测出基因的功能。针对这些问题,近年来出现了基因组上下文分析方法 (Genome context analysis),通过尽量发掘、综合基因组中蕴藏的相关重要信息,以准确预测基因的功能[2,13],并且通过实验加以验证,有效解决缺失基因问题,实现代谢途径的重构 (图 1)。以下将对代谢途径重构的各个步骤进行说明。
1.1.1 代谢途径的初步构建
基于微生物的完全基因组序列信息,利用SEED (http://theseed.uchicago.edu/FIG/index.cgi)、KEGG等公共数据库平台,在先验知识框架下初步构建代谢途径。系统地分析所涉及的各个酶、转运蛋白和调控蛋白,找到对应的编码基因;根据一条途径上各步反应的连续性,发现那些理应存在的代谢元件、但尚未找到对应基因的缺失基因问题。SEED中包含大量经专家分析注释过的代谢途径数据库,覆盖了大部分的微生物代谢网络,如包括枯草芽胞杆菌等模式微生物中几乎所有的生化和运输反应,此外还提供了一个基因组功能注释和分析的良好平台。
1.1.2 预测基因功能、填补代谢途径缺口
利用基因组上下文分析方法,结合基因簇[4,14-15]、结构域融合[16-17]、基因系统发育谱[18]以及调节位点共享[5,19]等多种重要的基因组信息,推断有关基因的功能关联性,进而准确预测基因的功能,有效地解决缺失基因问题、填补途径缺口,重构出完整的代谢途径 (图2)。

图1 基于比较基因组学重构代谢途径和调控网络Fig. 1 Comparative genomic reconstruction of regulatory and metabolic networks.
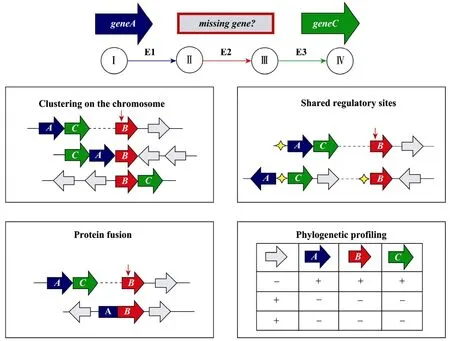
图2 基因组上下文分析技术Fig. 2 Techniques of genome context analysis.
基因簇——由于原核微生物染色体上功能相关的基因往往会聚集在一起,基因簇是推测不同基因功能耦合的重要依据。
结构域融合 (Protein fusion)——一个微生物中的两个不同基因在另一个微生物中融合成一个基因,预示着基因的功能相关。
基因系统发育谱 (Phylogenetic profiling)——基于同一个代谢途径上的基因在不同微生物中往往会同时存在或同时缺失,考察多个基因组上的基因存在规律可以提供功能耦合的依据。
调节位点共享 (Shared regulatory sites)——分析基因上游的调控区序列 (具体方法见1.2),找出共享调节位点的所有基因,即被一个调控因子所控制、同属一个调节单元 (Regulon) 的基因。基因的共调节是推测不同基因功能关联的重要依据。
对于上述比较基因组学研究产生的重要功能预测 (如新的功能基因),将用实验来加以验证。
1.2 调节单元 (Regulon) 重构
长期进化的结果使得微生物的代谢功能具有为自身服务的本能,其代谢过程处于最经济的状态,一般不会过量积累不必要的代谢产物。同时微生物能够根据外界环境条件的改变对自身的生理和代谢功能进行适当调整,以适应环境的变化。这种控制是在转录水平、翻译水平和酶活性等不同层面上调节的复杂过程,构成了一个精密的分子调控网络。细胞工厂要求微生物细胞能够定向、高效地生产人类所需要的目标化合物,这就必须去有目的地干扰、改变微生物原有的调控体系。如果不了解代谢途径的分子调控机制,就难以实现对细胞功能的理性调节。
细菌主要通过在转录水平和翻译水平上进行调节、改变相关基因的表达来适应外界环境的变化。其中转录因子 (包括全局调控因子和途径特异性调控因子) 具有非常重要的作用,它们通过特异性地结合顺式调控DNA序列 (转录因子结合位点) 来激活或抑制基因的转录过程。此外细菌中还有各种RNA调节系统 (如核开关 Riboswitch) 控制基因的表达。随着大量的原核微生物的基因组得以破译,基于这些序列信息利用比较基因组学技术预测顺式调节元件 (转录因子结合位点和 RNA元件),使得我们可以重构各个调节单元,即由一个转录因子或调节RNA调控的所有操纵子,在此基础上进而构建细胞内的转录调控网络[5]。这将为基因功能预测提供重要证据 (见1.1),同时将阐明对代谢途径起重要调控作用的转录因子以及代谢物和转录因子之间、不同转录因子之间的相互作用关系,从而显著提高对代谢调控的理解和认识,指导以基因操作来调控代谢途径及代谢功能。
然而,一个转录因子在不同基因上的结合位点具有一定的保守性,又不完全相同,这给识别转录因子结合位点 (Transcription factor-binding sites) 的工作带来了困难,使得预测结果普遍存在假阳性率偏高的问题。近年来出现了一些比较基因组学的新技术,通过发掘、分析基因的上游调控区序列,可以准确预测转录因子结合位点,实现调节单元的重构[5,20-22]。以下将对调节单元重构的各个步骤进行说明。
1.2.1 转录因子结合位点模型的建立
对于某一转录因子,分析哪些基因组上含有其编码基因;根据模式菌中受该转录因子调控的靶基因的信息,在一定进化距离内、并且含有该转录因子基因的一组基因组中找出靶基因的同源基因;结合代谢途径重构的结果 (见1.1),获得这些基因在基因组上的分布情况。分析这些基因集合的上游调控区序列,找出在多个基因上游出现的DNA回文序列或者串联的重复序列,即是可能的转录因子结合位点;选取信息容量最大的一组DNA序列,即其与转录因子结合的概率最大,构建其位置权重矩阵模型(Position weight matrix)[20]。
1.2.2 转录因子结合位点的识别、调节单元的重构
利用获得的位置权重矩阵模型在全基因组范围内进行搜索,找出更多的该转录因子的结合位点,获得相应的靶基因信息。在多物种间比较获得的靶基因,利用系统发育足迹分析法 (Phylogenetic footprinting) 去除其中的假阳性结果。这种方法是基于转录因子结合位点在进化上相对保守的基本假设,即由于转录因子结合位点有调控功能,在进化速度上要慢于其他没有功能的非编码序列[5,23]。通过有机结合“共调控”和“进化上保守”这两种信息,能够大幅提高转录因子结合位点和靶基因预测的准确性。这样获得在各个微生物中该转录因子所调控的基因集合,即调节单元。
对于上述比较基因组学研究产生的重要功能预测 (如新的转录因子与 DNA特异性结合序列),将用实验来加以验证。
1.3 实验验证
上述的比较基因组学研究将产生一组代谢途径上基因功能的预测,包括新的功能基因 (如酶、转运蛋白、调控蛋白的编码基因) 以及新的调控元件(如转录因子特异性结合的DNA序列)。这些基因功能预测可以通过传统的或高通量的实验技术来加以验证。
对于酶或转运蛋白等功能预测,可以通过传统的基因功能鉴定方法来进行验证。例如,对编码基因进行敲除,研究关键酶活性或转运能力对代谢功能的影响,分析该基因的生理学功能;考察功能基因能否在相应的模式菌突变株中实现功能互补,即该基因的表达能否使模式菌突变株缺失的功能得以恢复;提取粗酶液,或者分离纯化关键酶,测定相应的酶活性,分析该酶的生化学功能。
对于转录因子或调控元件等功能预测,可以通过传统的或高通量的实验技术来加以验证。例如,对转录因子的编码基因进行敲除,研究该转录因子的缺失对重要靶基因转录水平的影响;利用融合蛋白报告系统 (Reporter fusion) 分析转录因子在体内对靶基因调控区序列的作用;利用凝胶迁移率(Electrophoretic mobility shifts) 或DNase I足迹法(DNase I footprinting) 实验确认转录因子与预测的转录因子结合位点的结合情况。近年来也出现了一些高通量的转录组和蛋白质组的分析方法,如染色质免疫沉淀技术与基因芯片相结合的 ChIP-on-chip技术被越来越广泛地用于研究在全基因组上的转录因子结合位点[24]。
2 应用举例:丙酮丁醇梭菌中木糖代谢途径的重构
丙酮丁醇梭菌 Clostridium acetobutylicum是丁醇发酵工业的主要生产菌株,也是产溶剂梭菌研究中的模式菌。该菌能够利用木质纤维素中最主要的五碳糖——木糖,然而对于哪些基因编码木糖途径的关键酶和转运蛋白、以及这些基因表达的调控机制,目前还不清楚,从而难以通过代谢工程改造来提高丙酮丁醇梭菌的木糖代谢能力。
细菌中木糖的代谢大多是在木糖异构酶和木酮糖激酶的作用下生成5-磷酸-木酮糖,进入磷酸戊糖途径 (Pentose phosphate pathway)。然而,通过同源序列比对的方法在丙酮丁醇梭菌 ATCC824的基因组上无法找到编码木糖异构酶的同源基因,而且存在若干个编码木酮糖激酶的同源基因,同时丙酮丁醇梭菌中木糖代谢途径的调控机制仍不清楚。
针对上述问题,我们利用比较基因组学方法分析了梭菌纲和芽胞杆菌纲微生物的基因组序列信息,重构了它们的木糖和木寡聚糖利用的代谢途径和调节单元 (图 3)[25]。通过分析基因簇和调节位点共享,我们发现在丙酮丁醇梭菌及其他一些梭菌中存在一个新的编码木糖异构酶的基因,其与目前已知的木糖异构酶编码基因不具备同源性;预测了木糖代谢途径上的其他基因,包括编码木酮糖激酶、木糖转运蛋白、木糖调控因子的基因、以及木寡聚糖利用相关的基因。这些基因功能预测通过分子生物学和生物化学的实验技术在丙酮丁醇梭菌中进行了验证。此外,通过基因调控区域序列的比较分析,重构了木糖调控因子的调节单元,发现丙酮丁醇梭菌及其他一些梭菌中具有新的特异性结合木糖调控因子的DNA序列,其不同于目前已知的枯草芽胞杆菌中的木糖调控因子 DNA结合序列,该预测已通过实验得以证实。
3 结论与展望
本文对用于代谢途径和调控网络重构的一些新型比较基因组学技术进行了介绍,并举例说明了它们的应用。运用这些技术能够有效地解决缺失基因问题、准确地预测基因的功能,为实验分析提供指导,从而大大减轻实验工作量,提高工作效率。文中引用的一些成功实例表明,将这些新型比较基因组学技术与湿实验进行有机结合,能够显著提高基因功能注释和代谢网络重构的质量,并且发现新的代谢功能基因、调控元件、甚至新的代谢途径。本文的主要目的之一是希望实验工作者能够从这些成功范例中了解这类新型比较基因组学技术的重要性,从而利用这些技术来解决所关心的代谢途径中存在的缺失基因问题。
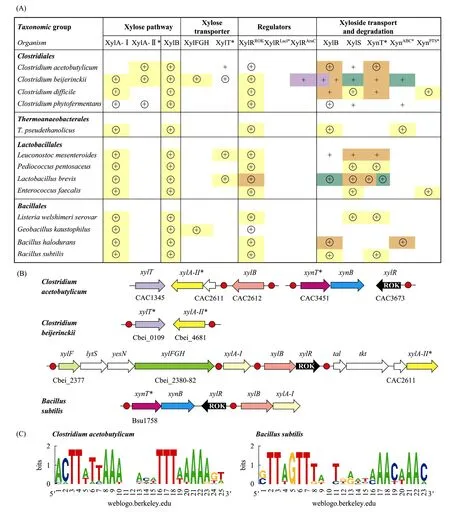
图3 厚壁菌门细菌木糖代谢途径的重构. (A) 木糖和木寡聚糖代谢相关基因的分布及特点;“+”表示基因存在;相同背景颜色表示基因在染色体上成簇存在;圆圈表示基因上游有转录因子XylR的DNA结合位点;(B) 基因组上下文分析;相同颜色的箭头表示同源基因;红色圆圈表示XylR的DNA结合位点;(C) 丙酮丁醇梭菌和枯草芽孢杆菌中的XylR特异性结合DNA序列Fig. 3 Reconstruction of xylose utilization pathway and regulons in Firmicutes. (A) Occurrence and features of genes involved in xylose and xyloside utilization pathway. The presence of genes for the respective functional roles is shown by “+”. Genes clustered on the chromosome are marked by the same background color. Candidate XylR regulon members are circled. (B) Genomic context analysis. Homologous genes are marked by matching colors. Candidate regulatory sites of XylR are shown by red circles. (C) DNA recognition motifs of XylR from Clostridium acetobutylicum and Bacillus subtilis.
随着多种原核生物的全基因序列得以破译,这些大量的信息为基因组上下文分析技术的应用提供了很好的数据背景。用于分析的基因组的数目和多样性往往决定了这些技术能否得以成功应用。例如,在一组非常相近的基因组上 (如同一种类的不同菌株) 基因间的序列往往也是相同的,那么这些基因组就不能用于转录因子结合位点的预测分析;另一方面,如果用于分析的微生物的亲缘关系相差很远(如革兰氏阳性厚壁菌门细菌和革兰氏阴性变形菌门细菌),调控元件在这些基因组上并不保守,那么也很难得到准确的转录因子结合位点模型。虽然基因簇、调节位点共享和基因系统发育谱分析只适用于原核生物的基因组,但是大量的原核生物基因组信息显示,大多数的真核生物中代谢酶都可以在一组原核生物中找到功能对等的蛋白质。因此,这些新型比较基因组学技术也将间接地为真核生物基因组以及元基因组数据的准确功能注释提供重要的帮助。
REFERENCES
[1] Overbeek R, Begley T, Butler RM, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res, 2005, 33: 5691−5702.
[2] Osterman A, Overbeek R. Missing genes in metabolic pathways: a comparative genomics approach. Curr Opin Chem Biol, 2003, 7: 238−251.
[3] Koonin EV, Galperin MY. Sequence–Evolution–Function. Computational Approaches in Comparative Genomics. Boston: Kluwer Academic Publishers, 2002.
[4] Overbeek R, Fonstein M, D’Souza M, et al. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA, 1999, 96: 2896−2901.
[5] Rodionov DA. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem Rev, 2007, 107: 3467−3497.
[6] Pinchuk GE, Rodionov DA, Yang C, et al. Genomic reconstruction of Shewanella oneidensis MR-1 metabolism reveals a previously uncharacterized machinery for lactate utilization. Proc Natl Acad Sci USA, 2009, 106: 2874−2879.
[7] Yang C, Rodionov DA, Li X, et al. Comparative genomics and experimental characterization of N-acetylglucosamine utilization pathway of Shewanella oneidensis. J Biol Chem, 2006, 281: 29872−29885.
[8] Yang C, Rodionov DA, Rodionova IA, et al. Glycerate 2-kinase of Thermotoga maritima and genomic reconstruction of related metabolic pathways. J Bacteriol, 2008, 190: 1773−1782.
[9] Rodionov DA, Kurnasov OV, Stec B, et al. Genome identification and in vitro reconstitution of a complete biosynthetic pathway for the osmolyte di-myo-inositolphosphate. Proc Natl Acad Sci USA, 2007, 104: 4279−4284.
[10] Sorci L, Martynowski D, Rodionov DA, et al. Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. Proc Natl Acad Sci USA, 2009, 106: 3083−3088.
[11] Piskur J, Schnackerz KD, Andersen G, et al. Comparative genomics reveals novel biochemical pathways. Trends Genet, 2007, 23: 369−372.
[12] Rodionov DA, Gelfand MS. Comparative genomics and functional annotation of bacterial transporters. Phys Life Rev, 2008, 5: 22−49.
[13] Wolf YI, Rogozin IB, Kondrashov AS, et al. Genome alignment, evolution of prokaryotic genome organization, and prediction of gene function using genomic context. Genome Res, 2001, 11: 356−372.
[14] Galperin MY, Koonin EV. Whos’s your neighbor? New computational approaches for functional genomics. Nat Biotechnol, 2000, 18: 609−613.
[15] Snel B, Bork P, Huynen MA. The identification of functional modules from the genomic association of genes. Proc Natl Acad Sci USA, 2002, 99: 5890−5895.
[16] Enright AJ, Iiopoulos I, Kyrpides NC, et al. Protein interaction maps for complete genomes based on gene fusion events. Nature, 1999, 402: 86−90.
[17] Marcotte EM, Pellegrini M, Ng HL, et al. Detecting protein function and protein-protein interactions from genome sequences. Science, 1999, 285: 751−753.
[18] Pellegrini M, Marcotte EM, Thompson MJ, et al. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA, 1999, 96: 4285−4288.
[19] Manson McGuire A, Church GM. Predicting regulons and their cis-regulatory motifs by comparative genomics. Nucleic Acids Res, 2000, 28: 4523−4530.
[20] Gelfand MS, Koonin EV, Mironov AA. Prediction of transcription regulatory sites in Archaea by a comparative genomic approach. Nucleic Acids Res, 2000, 28: 695−705.
[21] Tan K, McCue LA, Stormo GD. Making connections between novel transcription factors and their DNA motifs. Genome Res, 2005, 15: 312−320.
[22] Tan K, Moreno-Hagelsieb G, Collado-Vides J, et al. A comparative genomics approach to prediction of new members of regulons. Genome Res, 2001, 11: 566−584.
[23] Blanchette M, Tompa M. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res, 2002, 12: 739−748.
[24] Grainger DC, Lee DJ, Busby SJ. Direct methods for studying transcription regulatory proteins and RNA polymerase in bacteria. Curr Opin Microbiol, 2009, 12: 531−535.
[25] Gu Y, Ding Y, Ren C, et al. Reconstruction of xylose utilization pathway and regulons in Firmicutes. BMC Genomics, 2010, 11: 255.
Comparative genomic reconstruction of regulatory and metabolic networks in bacteria
Chen Yang
Key Laboratory of Synthetic Biology, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China
A large and growing number of complete genomes from diverse species open tremendous opportunities for getting deep insights into cell metabolism. This increased understanding strongly supports engineering of cell metabolism for microbial production. In spite of the recent progress, a large fraction of genes in most of the available genomes remain incorrectly or imprecisely annotated. In this paper we review some of the new comparative genomics techniques used to reconstruct regulatory and metabolic networks from genomic data, reveal gaps in current knowledge, and identify previously uncharacterized genes. The application will be discussed by using a recent example–reconstruction of xylose utilization pathway in Clostridium acetobutylicum.
comparative genomics, reconstruction of metabolic pathways, reconstruction of transcriptional regulatory networks, genome context analysis
自上个世纪末以来微生物基因组学的发展突飞猛进,迄今为止国际上已经公布了超过 1 000组完整的原核微生物基因组序列 (http://www.ncbi.nlm. nih.gov/genomes/lproks.cgi)。这为从功能基因与蛋白、网络及其调控等不同的角度,全面理解与认识微生物的代谢过程、构建细胞工厂,提供了丰富的背景信息[1]。基于基因组序列进行代谢网络重构,不仅能够发掘模式菌的新的代谢特性,而且能够预测我们了解甚少的某些微生物的代谢潜能,发现新的代谢功能基因、调控元件、以及新的代谢途径[2]。这些都有助于我们优化设计细胞内从原料到产品的生物合成路线,重新组装部分代谢和调控元件,进而构建具有定向的转化和合成能力的细胞工厂。
June 4, 2010; Accepted: August 11, 2010
Supported by:One-hundred People Program of Chinese Academy of Sciences (No. KSCX2-YW-G-029), National Basic Research Program of China (973 Program) (No. 2007CB707802).
Chen Yang. Tel: +86-21-54924152; E-mail: chenyang@sibs.ac.cn
中国科学院项目百人计划 (No. KSCX2-YW-G-029),国家重点基础研究发展计划 (973计划) (No. 2007CB707802) 资助。
