基因组尺度代谢网络研究进展
2010-02-09王晖马红武赵学明
王晖,马红武,赵学明
1 天津大学化工学院生物工程系,天津 300072
2 教育部系统生物工程重点实验室,天津 300072
3 天津大学-爱丁堡大学系统生物学与合成生物学联合研究中心,天津 300072
基因组尺度代谢网络研究进展
王晖1,2,3,马红武1,2,3,赵学明1,2,3
1 天津大学化工学院生物工程系,天津 300072
2 教育部系统生物工程重点实验室,天津 300072
3 天津大学-爱丁堡大学系统生物学与合成生物学联合研究中心,天津 300072
基因组尺度代谢网络从基因组序列出发,结合基因、蛋白质、代谢数据库和实验数据,从系统的角度定量研究生命体的代谢过程,了解各个组分之间的相互作用关系。这类网络模型对于生命活动理论研究和优良工程菌的构建都具有重要的理论和实践意义。以下结合作者的实际研究经验,对基因组尺度代谢网络从重构到模拟直至应用进行了较为详细的介绍,并讨论了一些目前存在的难题和未来的研究方向。
基因组尺度,代谢网络,系统生物学,代谢工程
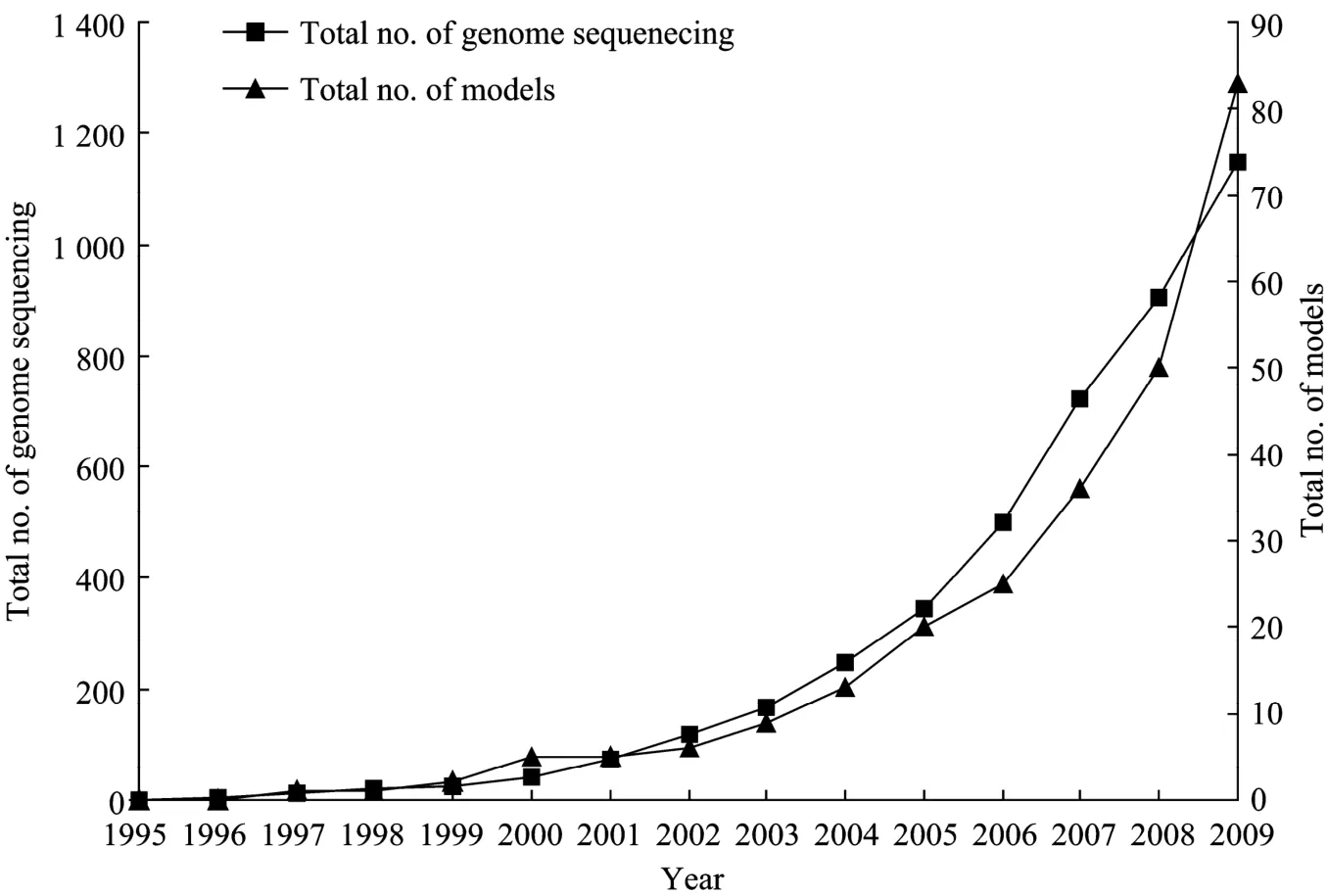
图1 已测序的物种和已经构建的基因组尺度代谢网络模型数目Fig. 1 The number of sequenced species and reconstructed genome-scale metabolic networks.
基因组尺度代谢网络是系统生物学的重要工具,通过结合计算机模型和实验数据,从系统角度分析复杂的生物系统。这类模型主要应用在5个方面[5]:与高通量技术相结合,更有效地分析处理高通量数据;指导代谢工程;基于假设指导有目的性地发现研究;探索物种间的相互关系;网络特性的分析和研究。基因组尺度代谢网络模型作为工具,无论在生物体以及生命活动的理论研究上,还是在指导代谢工程进行工程菌改造上,都具有非常重要的理论和实践意义。
国外已经有数篇关于基因组尺度代谢网络模型构建以及模拟方法的综述[6-10],在这里作者结合自己的研究经历,较为详细地阐述模型的重构过程,模拟方法以及应用。
1 基因组尺度代谢网络重构
基因组尺度代谢网络模型的重构是个循环往复的过程,主要包括3个步骤:代谢网络数据库的建立、数学模型的建立和模拟运算验证模型。通过模拟反复循环验证,当模拟结果的准确率达到一定水平后,网络构建也就完成了,可以进行其他预测等工作。
1.1 基因组尺度代谢网络数据库的建立
基因组尺度代谢网络数据库跟通常意义的数据库不一样,建立这样的数据库就是从生物信息数据库和文献中提取出需要的数据,在电脑中进行整理和精炼。建立这样一个数据库通常需要 3个步骤:数据收集、关系模型建立和数据整理。
1.1.1 数据收集
基因组尺度代谢网络重构的主要数据来源来自各种生物信息数据库,随着计算机和互联网络的发展,大量的生物学信息可以从各大数据库中免费获得,这些数据库包括基因组数据库、蛋白质数据库以及一些代谢反应数据库、表 1中列出了一些常用的数据库。
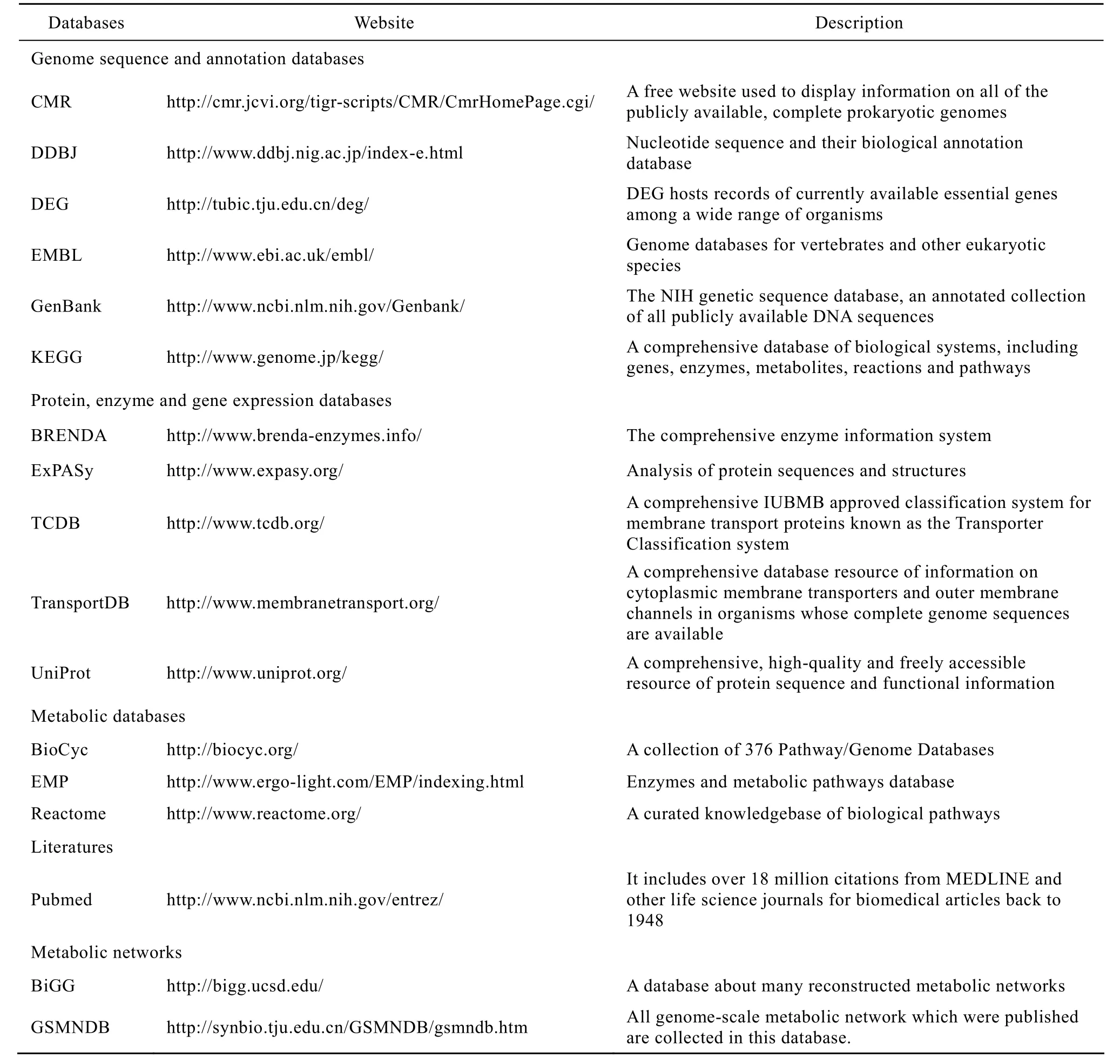
表1 基因组尺度代谢网络构建常用的数据库Table 1 Databases frequently used for reconstruction of genome-scale metabolic network
以上提到的数据库大都提供批量下载,下载数据完毕后通过VBA等编程语言,将所需的数据提取出来,对于不提供批量下载的数据库,也可以通过Python等语言直接从网页提取数据。提取得到的数据放在Excel表中,由于Excel是专用的表格类数据处理软件,因此将数据放入 Excel表中进行整理和精炼十分便利。
需要提取和用到的原始数据主要有:物种特异的基因、蛋白质、反应和代谢物信息。然而单一数据库提供的数据往往是有限的,而且各个数据库之间由于注释算法和其他组织结构不同,可能会导致数据的不一致性,因此通常构建基因组尺度代谢网络的过程中,原始数据都是来自多个数据库的。
另一个原始数据的重要来源是大量的文献和书籍。需要使用文献搜索引擎广泛地搜索来自文献和书籍的信息,为了数据收集得全面,通常要使用几个文献搜索引擎并用进行搜索,同时关键字也要交叉组合进行搜索以确保不会遗漏信息。因为很多近期的文献中可能提供了新基因功能注释等与网络重构密切相关的信息,而这些信息在数据库中收录会比较慢,所以即便通过文献和书籍添加的信息量会比较小,但是这部分信息也是十分重要的,而且可靠性最高。
1.1.2 关系模型的建立
关系模型就是将上一步提取到的各种数据关联在一起,如基因与酶、酶与反应、反应与代谢物之间的关联。
基因与反应的对应关系是通过酶蛋白进行介导的,即基因通过注释得到基因编码的蛋白信息,通常与代谢活动密切相关的蛋白都是酶,酶都会对应一定的酶号,通过酶号关联该酶催化的反应,通过这种方式可以得到基因与反应的对应关系。通过反应的代谢方程式,将代谢物关联在一起从而构成整个代谢网络。
1.1.3 数据整理
数据整理是基因组尺度代谢网络构建过程中最费时费力的一个环节,因为前面步骤提取得到的数据往往会存在很多问题,例如:生物信息数据库中往往不会提供物种特异性的信息,即某些代谢反应在物种中不会发生,而此类反应生物信息数据库中往往没有特别标注,这就需要在数据整理中将不会在物种内发生的反应剔除。另外,原始数据中可能会存在一些错误数据,还需要结合大量文献进行佐证,这些都需要大量的时间来进行人工校正。
前面提到用于构建代谢网络的原始数据来源于不同的数据库,因此就要对不同来源的数据进行比对精炼。一般都是通过不同数据库之间的 ID Mapping,使不同数据库来源的同一基因对应的信息相互关联。对于注释信息如酶号不一致的基因,应该通过查阅文献或参考其他数据库进一步确认。同样,对于不同来源的代谢反应信息 (如 KEGG和BioCyc),也应该进行比对,以确保反应方程式和反应方向等信息准确。
从数据库得到的原始数据的有效性是不同的,因此对数据进行可靠性分级也是必要的。通常情况下,有确凿的文献和实验验证的数据,是最可靠的;如果多个数据库得到的数据完全一致,这样的数据可靠性较高;单一数据库得到的数据可靠性一般;单纯依靠注释信息得到的基因功能信息,可靠性最低。
大分子的合成与修饰反应往往需要特别的处理。蛋白质、DNA、RNA、肽聚糖和磷壁酸等大分子的具体合成过程很复杂,而且种类繁多,很多合成修饰机制还没有完全了解。因此对于构建基因组尺度代谢网络来说,通常都是将这些大分子的合成反应按照一定的权重归并到生物量合成反应中。
反应数据的整理。反应数据的整理涉及反应方程式的确定、反应方向的确定、反应辅酶的确定、反应质量以及电荷配平等。通常数据库中存在大量的反应冗余、反应方向不确定等问题,这些都需要细致的人工校正来进行确认。人工校正工作可以采用多种方法或者多个数据来源来综合评定,例如反应方向性的确定,可以参考文献资料以及相关教科书,也可以参考KEGG Pathway和Brenda等数据库,还可以通过热力学以及拓扑结构等来进行确认,如果实在找不到相关依据,往往采用经验规则[10]。其他反应信息确定也可以采用类似的方法,综合多种信息来源来进行最终确认。
分析网络断口 (Gap)。在代谢网络中会存在一些代谢物只有消耗没有生成,或者只有生成没有消耗,这些代谢物通常称为末端代谢物 (Dead ends)。这些末端代谢物可以通过编程进行提取和识别。这类代谢物的产生通常是由于信息量不够,或者我们对物种的了解不足,有些应该存在的反应在我们收集数据的过程中没有找到,这样就需要更广泛的查阅文献,寻找相关的信息进行补充,如果实在无法找到确凿的信息进行验证,可以在代谢网络中添加demand reactions[10]来解决。
对于较为复杂的物种,通常还要设定合适的分室信息,例如真核生物,要将各个主要细胞器例如线粒体、过氧化物酶体等细胞器作为单独分室进行处理。
通过上述提取整理过程后,我们得到的结果是一个反应列表 (通常保存在 Excel表格中),包括了物种的基因-蛋白质-反应对应信息以及反应的详细信息,包括:反应方程式、反应方向、反应所属途径和所属分室等。
1.2 数学模型的建立
反应列表整理完成后,我们要将其转化为数学模型才可以在计算机上进行相应模拟。基因组尺度代谢网络模型的核心就是计量系数矩阵。
计量系数矩阵是将前面得到的反应列表中各个反应方程式代谢物的系数汇总在一起构成的一个多维矩阵。通常用S表示,在S中每一行对应一个代谢物,每一列对应一个反应。
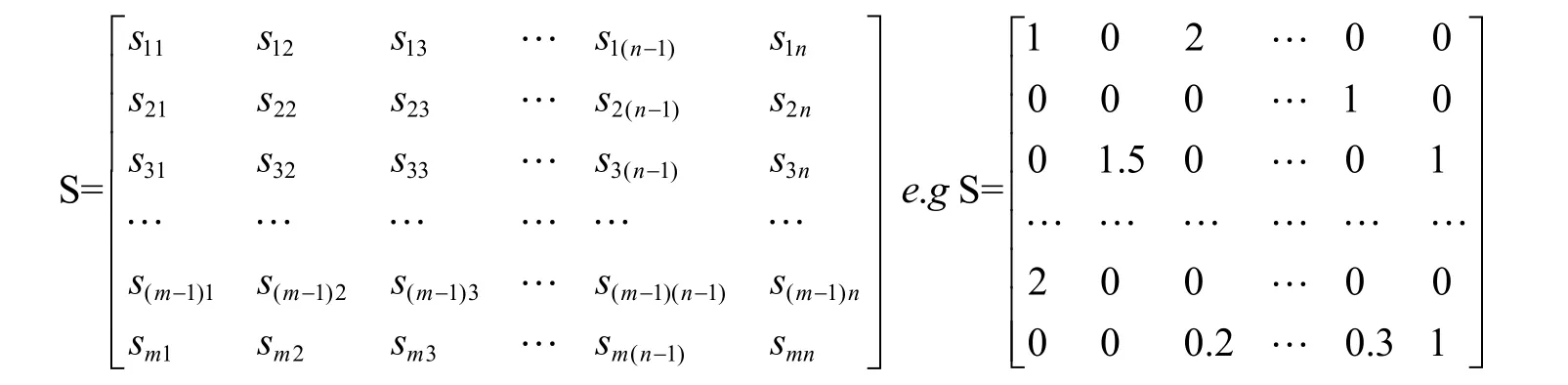
计量系数矩阵中还包括其他 2个主要部分,首先是生物量组成。生物量的组成通过文献查找各个组分的含量获得,如果实在找不到物种的确切组成,可以借鉴其他相近的物种,然后根据具体含量定量作为系数,组成生物量合成方程式,并将系数等信息合并入计量系数矩阵。其次是运输反应,根据生物可以利用的底物和分泌的产物添加运输反应,一部分可以由基因组注释得到膜运输蛋白,从而可以确定运输反应的存在,另外一部分先通过查找文献,确定菌体的基础合成培养基,然后根据需要添加相应的运输反应。同时还需要为所有的胞外代谢物添加交换反应 (Exchange reaction),交换反应可以理解为胞外代谢物向细胞膜的扩散。这些运输反应也表示为反应方程式形式,都要汇总进计量系数矩阵中。
计量系数矩阵可以通过 VBA编程,直接将Excel中的反应方程式转化为 SBML (Systems Biology Markup Language)[11]格式的计量系数矩阵。SBML的数据文件是通用的系统生物学语言格式的文件,可以被大多数生物模拟软件识别并加载。
1.3 模拟运算
目前常用的基因组尺度代谢网络的模拟算法都是基于约束的优化模拟方法[8],其中以线性优化为主,最基本的组成部分就是:约束条件、决策变量和目标函数。而最常用的算法就是通量平衡分析(Flux Balance Analysis:FBA),该算法假设系统处于拟稳态,即中间代谢物的生成与消耗相同,在这种假设下设置一定的约束条件和目标函数,来研究胞内相应状态下的通量分布情况。其基本算法可以由以下数学表达式来表示:
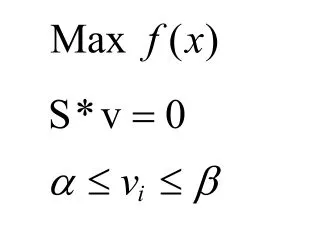
其中f(x) 表示目标函数,S表示计量系数矩阵,v表示各步反应的通量,β和α分别表示反应通量的上下限。目标函数可以根据研究需要自行设定,可以设定为生物量,即表示生物量积累最多情况下的通量分布,也可以设定为某种产品的产量,从而研究目标产品生产最大化或者副产物最小化等状态下代谢通量的分布。
上面只是通用的FBA算法,以通用的算法为基础,还有很多针对特定问题的算法[8],用来解决特殊研究对象的优化模拟。
模拟运算的计算平台有很多,其中Matlab®(The MathworksTM) 使用得最为广泛。在Matlab中,通过调用Matlab的内置函数对计量系数矩阵进行操作,对于熟练使用Matlab的研究者,可以直接使用Matlab的字符串函数,通过指定字符串来操作矩阵;对于不熟悉 Matlab的研究者,可以通过之前编排的序号来指定矩阵中的相应元素。对于最优化计算,可以使用Matlab的最优化工具箱,最优化工具箱中有各种函数,可以方便地用于解决各种优化问题 (线性、非线性、多目标优化和二次规划等)。在Matlab平台的基础上,现在开发出一些专门用于基于约束的模拟工具,如COBRA (Constraints Based Reconstruction and Analysis) 工具箱[12]。COBRA 工具箱是由一系列针对基于约束的构建和模拟用途而编写的函数组成,含有可以读取SBML或Excel格式的模型的函数,使用者不再需要自己编写提取矩阵的程序。具体用法可以参阅文献[10]。
通过模拟的结果和实验结果相比对,我们可以完善得到的模型。对于结果不一致的,一方面可能是模型不够完善导致的结果差异;另一方面,模拟的结果可能揭示了一些我们尚未研究到的内容。因此模拟验证、修正模型、修正后再模拟验证是个循环往复的过程,直到模拟结果的准确率满足要求后,一个物种的基因组尺度代谢网络模型才算最终构建完成。
2 基因组尺度代谢网络的应用
基因组尺度代谢网络构建完成后,可以用于菌种改进、发现药物靶点、代谢工程操作靶点识别、生长表型预测等多种用途,这里简单介绍此类模型应用最广泛的几个方面。
2.1 基因敲除研究
基因敲除研究是基因组尺度代谢网络模拟预测最突出的应用之一。即使现在高通量技术发展得很快,试验中批量确定基因的必需性仍然是费时费力的,而且成本很高。然而对于基因组尺度代谢网络模型来说,基因敲除通过基因-蛋白-反应相互关系转化为反应的敲除,目标函数设定为生物量最大化,将基因对应的反应通量人为设定成 0即可实现。使用这种方法可以快捷方便地同时检测数百个基因是否是必需基因,虽然结果无法保证百分之百正确,但是此种模拟的算法和相关技术都已经很成熟,准确率基本上都可以达到80%以上,因此已经基本上成为构建基因组尺度代谢网络必做的模拟工作[13-15]。
模拟得到的必需基因结果与实验结果进行比对也是验证模型精确率的一个重要方法,然而由于批量实验验证必需基因难度不小,目前很多物种的必需基因都没有得到实验验证,此类验证模型的方法也无法实施。但是对于一些常用的物种如大肠杆菌、枯草芽胞杆菌、酿酒酵母等有很多必需基因已经通过实验证实 (DEG[16]:http://tubic.tju.edu.cn/deg/),对于这些物种来说,此项模拟结果的精确度往往是一个基因组尺度代谢网络模型质量高低的标准。干、湿实验比对结果又可以分为 4类[17]:TP (True Positive实验和模拟结果都为正)、TN (True Negative实验和模拟结果都为负)、FP (False Positive实验为必需基因,模拟为非必需基因) 和 FN (False Negative实验为非必需基因,模拟为必需基因)。其中FP和FN往往能提供模型完善的重要靶点,产生这些差异结果的主要原因有:基因组尺度代谢网络缺乏调控机制;没有考虑物种内的拓扑结构;网络中存在断口;目标函数中的组分不全;培养基组分设定不合理;没有考虑胞内代谢物过量积累的毒素效应;缺乏一些胞内非代谢过程;错误的基因注释;实验数据不准确等等。这些原因会提供大量改进模型和设计实验的靶点。
基因必需性研究并不是使用基因组尺度网络进行基因敲除研究的唯一目的,通过基因敲除还可以研究生物的一些特殊生理过程的机理[18]。
2.2 发现药物靶点
基因组尺度代谢网络可以用于药物研发,包括药物靶点识别、抗菌药物的研发和疫苗的改良。其中人类[4,19]和致病菌[20-22]的基因组尺度代谢网络尤为重要,因为很多人类疾病都与人的代谢紊乱以及致病菌的代谢机制密切相关。通过使用此类模型进行模拟研究,可以深入了解人在病态下的代谢状态,从而可以有针对性的采用一些医疗措施,对药物的研发也有很大帮助。
2.3 菌体改进和代谢工程
由于生物体中网络的刚性和冗余性,单一基因的改造往往得不到预期的效果。而基因组尺度代谢网络考虑生物体整体代谢,可以从更大范围内了解代谢过程和一些基因操作后的效果,因此对于优良工程菌的构建以及代谢工程操作都具有重大的意义。迄今为止,已经有很多研究通过基因组尺度代谢网络指导进行基因操作和工程菌构建,生产生物能源、生物基化学品及高附加值产品,表2给出了部分实例。
3 基因组尺度代谢网络研究展望
近几年,基因组尺度代谢网络模型发展非常迅速,不仅很多模式菌的基因组尺度代谢网络构建完成,而且相关技术、算法、各种模拟画图软件都有了很大的进步。然而目前仍然存在一些关键的问题暂时无法解决。
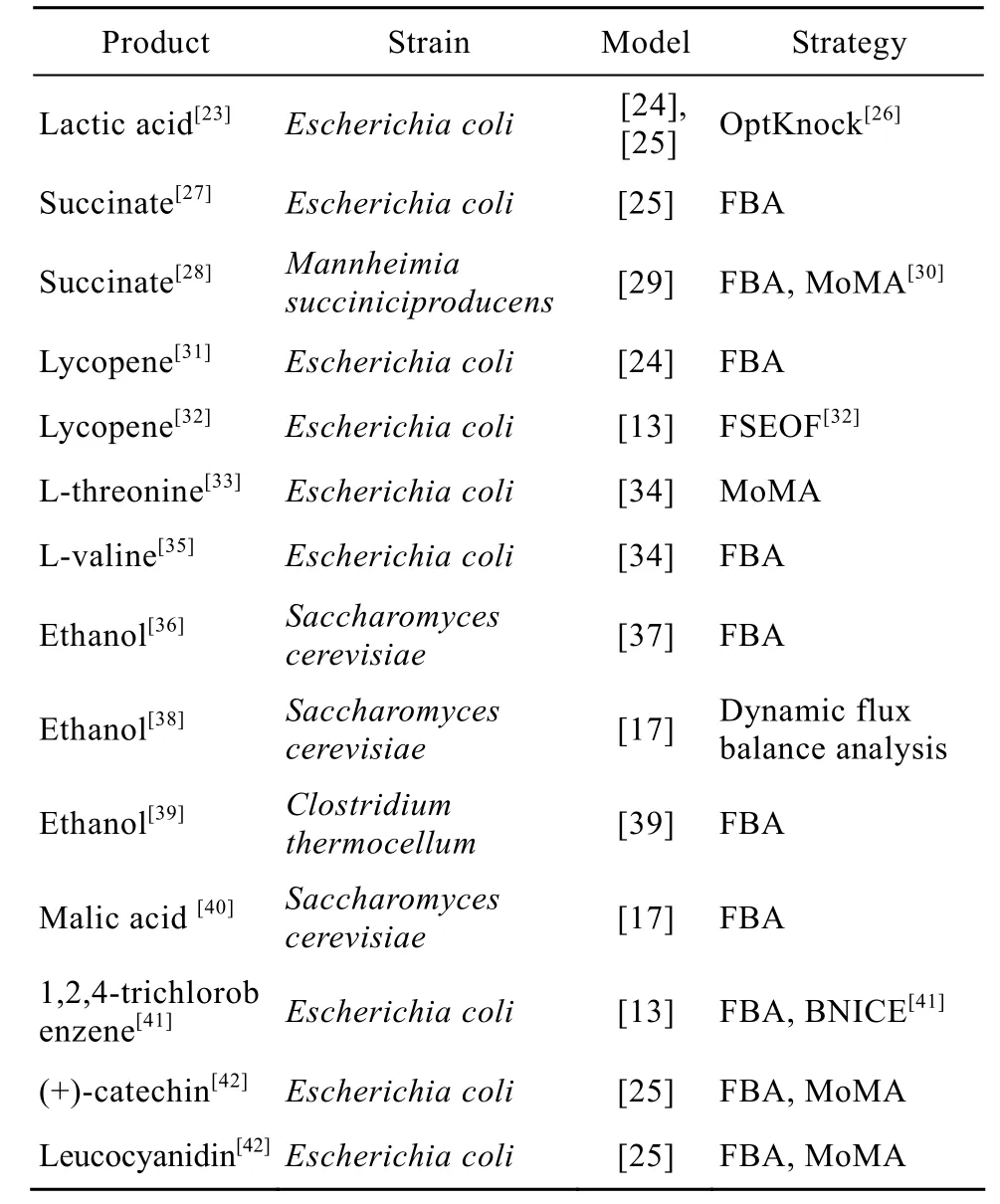
表2 应用基因组尺度代谢网络改良工程菌Table 2 Engineered strain improvement using genomescale metabolic network
首先,基因组尺度代谢网络与其他生物过程结合很少。在生物体中还有其他很多重要的机制,例如转录调控、信号转导等,虽然已经出现了基因组尺度的转录调控网络[43]和信号转导网络[44]计算机模型,但是目前仍然无法广泛的实践。如何将代谢网络、转录调控网络、信号转导网络等结合起来构成“大生物网络”[45]将成为研究热点。
其次,很多基因组尺度代谢网络重构需要的数据都不完善。目前很多生物信息数据库中的信息都存在一定量的错误和冗余,如何甄别这些无效信息成为很大的难点,因为这个要花费大量的时间和人力。此外,很多反应的方向无法确定和相关文献信息量不足也导致基因组尺度代谢网络的模拟结果必定存在误差。
再次,基因组尺度代谢网络的模拟大多都是基于拟稳态假设,而实际在生物体内,代谢物生成和消耗都是动态的,虽然目前已经有人尝试进行了动态FBA[46],但是如何广泛应用于基因组尺度的代谢网络,如何反映生物体实时状态下的代谢情况目前都是研究的难点。
最后,由于各个研究组所构建的基因组尺度代谢网络大多使用自己的编号以及数据整理格式,这些格式通常不统一,这对于模型的广泛应用以及不同研究组相互合作造成了不小的障碍。虽然2008年几个研究组进行了一些酿酒酵母基因组尺度代谢网络模型标准化的尝试[47],但是这些标准仍然没有广泛应用。作者所在的研究组搭建了世界上第一个收录全部基因组尺度代谢网络模型的数据库GSMNDB,并对所有已经发表的模型进行整理,努力做到格式和表示方式标准化,以便更多的人可以通过已经构建的模型,更快更深入地进入到该研究领域。
REFERENCES
[1] Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science, 1995, 269(5223): 496–512.
[2] Genomes OnLine Database (GOLD). [2010-06-04]. http://www.genomesonline.org/cgi-bin/GOLD/bin/gold.cgi.
[3] Gerlt JA, Babbitt PC. Can sequence determine function? Genome Biol, 2000, 1(5): REVIEWS0005.
[4] Ma HW, Sorokin A, Mazein A, et al. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol Syst Biol, 2007, 3: 135.
[5] Oberhardt MA, Palsson BØ, Papin JA. Applications of genome-scale metabolic reconstructions. Mol Syst Biol, 2009, 5: 320.
[6] Durot M, Bourguignon PY, Schachter V. Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol Rev, 2009, 33(1): 164−190.
[7] Feist AM, Herrgård MJ, Thiele I, et al. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol, 2009, 7(2): 129−143.
[8] Park JM, Kim TY, Lee SY. Constraints-based genome-scale metabolic simulation for systems metabolic engineering. Biotechnol Adv, 2009, 27(6): 979−988.
[9] Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc, 2010, 5(1): 93−121.
[10] Orth JD, Thiele I, Palsson BØ. What is flux balance analysis? Nat Biotechnol, 2010, 28(3): 245−248.
[11] Hucka M, Finney A, Bornstein BJ, et al. Evolving a lingua franca and associated software infrastructure for computational systems biology: the Systems Biology Markup Language (SBML) project. Syst Biol (Stevenage), 2004, 1(1): 41−53.
[12] Becker SA, Feist AM, Mo ML, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc, 2007, 2(3): 727−738.
[13] Feist AM, Henry CS, Reed JL, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol, 2007, 3: 121.
[14] David H, Ozçelik IS, Hofmann G, et al. Analysis of Aspergillus nidulans metabolism at the genome-scale. BMC Genomics, 2008, 9: 163.
[15] Oberhardt MA, Puchałka J, Fryer KE, et al. Genome-scale metabolic network analysis of the opportunistic pathogen Pseudomonas aeruginosa PAO1. J Bacteriol, 2008, 190(8): 2790−2803.
[16] Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res, 2009, 37(Database issue): D455−458.
[17] Duarte NC, Herrgård MJ, Palsson BØ. Reconstruction and validation of Saccharomyces cerevisiaei iND750, a fully compartmentalized genome-scale metabolic model. Genome Res, 2004, 14(7): 1298−1309.
[18] Resendis-Antonio O, Reed JL, Encarnación S, et al. Metabolic reconstruction and modeling of nitrogen fixation in Rhizobium etli. PLoS Comput Biol, 2007, 3(10): 1887−1895.
[19] Duarte NC, Becker SA, Jamshidi N, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA, 2007, 104(6): 1777−1782.
[20] Baart GJ, Zomer B, de Haan A, et al. Modeling Neisseria meningitidis metabolism: from genome to metabolic fluxes. Genome Biol, 2007, 8: R136.
[21] Beste DJ, Hooper T, Stewart G, et al. GSMN-TB: a web-based genome-scale network model of Mycobacterium tuberculosis metabolism. Genome Biol, 2007, 8: R89.
[22] Jamshidi N, Palsson BØ. Investigating the metabolic capabilities of Mycobacterium tuberculosis H37Rv using the in silico strain iNJ661 and proposing alternative drug targets. BMC Syst Biol, 2007, 1: 26.
[23] Fong SS, Burgard AP, Herring CD, et al. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol Bioeng, 2005, 91(5): 643−648.
[24] Edwards JS, Palsson BO. The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc Natl Acad Sci USA, 2000, 97(10): 5528−5533.
[25] Reed JL, Vo TD, Schilling CH, et al. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol, 2003, 4(9): R54.
[26] Burgard AP, Pharkya P, Maranas CD. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng, 2003, 84(6): 647−657.
[27] Wang QZ, Chen X, Yang YD, et al. Genome-scale in silico aided metabolic analysis and flux comparisons of Escherichia coli to improve succinate production. Appl Microbiol Biotechnol, 2006, 73(4): 887−894.
[28] Lee SY, Kim JM, Song H, et al. From genome sequence to integrated bioprocess for succinic acid production by Mannheimia succiniciproducens. Appl Microbiol Biotechnol, 2008, 79(1): 11−22.
[29] Kim TY, Kim HU, Park JM, et al. Genome-scale analysis of Mannheimia succiniciproducens metabolism. Biotechnol Bioeng, 2007, 97(4): 657−671.
[30] Segrè D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci USA, 2002, 99(23): 15112−15117.
[31] Alper H, Jin YS, Moxley JF, et al. Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab Eng, 2005, 7(3): 155−164.
[32] Choi HS, Lee SY, Kim TY, et al. In silico identification of gene amplification targets for improvement of lycopene production. Appl Environ Microbiol, 2010, 76(10): 3097−3105.
[33] Lee KH, Park JH, Kim TY, et al. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol Syst Biol, 2007, 3: 149.
[34] Lee SY, Woo HM, Lee DY, et al. Systems-level analysis of genome-scale in silico metabolic models using MetaFluxNet. Biotechnol Bioproc Eng, 2005, 10: 425−431.
[35] Park JH, Lee KH, Kim TY, et al. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc Natl Acad Sci USA, 2007, 104(19): 7797−7802.
[36] Bro C, Regenberg B, Förster J, et al. In silico aided metabolic engineering of Saccharomyces cerevisiae for improved bioethanol production. Metab Eng, 2006, 8(2): 102−111.
[37] Förster J, Famili I, Fu P, et al. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res, 2003, 13(2): 244−253.
[38] Hjersted JL, Henson MA, Mahadevan R. Genome-scale analysis of Saccharomyces cerevisiae metabolism and ethanol production in fed-batch culture. Biotechnol Bioeng, 2007, 97(5): 1190−1204.
[39] Roberts SB, Gowen CM, Brooks JP, et al. Genome-scale metabolic analysis of Clostridium thermocellum for bioethanol production. BMC Syst Biol, 2010, 4: 31.
[40] Zelle RM, de Hulster E, van Winden WA, et al. Malic acid production by Saccharomyces cerevisiae: engineering of pyruvate carboxylation, oxaloacetate reduction, and malate export. Appl Environ Microbiol, 2008, 74(9): 2766−2777.
[41] Finley SD, Broadbelt LJ, Hatzimanikatis V. In silico feasibility of novel biodegradation pathways for 1,2,4-trichlorobenzene. BMC Syst Biol, 2010, 4: 7.
[42] Chemler JA, Fowler ZL, McHugh KP, et al. Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab Eng, 2010, 12(2): 96−104.
[43] Thiele I, Jamshidi N, Fleming RM, et al. Genome-scale reconstruction of Escherichia coli's transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization. PLoS Comput Biol, 2009, 5(3): e1000312.
[44] Hyduke DR, Palsson BO. Towards genome-scale signalling-network reconstructions. Nat Rev Genet, 2010, 11(4): 297−307.
[45] Reed JL, Famili I, Thiele I, et al. Towards multidimensional genome annotation. Nat Rev Genet, 2006, 7(2): 130−141.
[46] Oddone GM, Mills DA, Block DE. A dynamic, genome-scale flux model of Lactococcus lactis to increase specific recombinant protein expression. Metab Eng, 2009, 11(6): 367−381.
[47] Herrgård MJ, Swainston N, Dobson P, et al. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat Biotechnol, 2008, 26(10): 1155−1160.
Progress in genome-scale metabolic network: a review
Hui Wang1,2,3, Hongwu Ma1,2,3, and Xueming Zhao1,2,3
1 Department of Biochemical Engineering, School of Chemical Engineering & Technology, Tianjin University, Tianjin 300072, China
2 Key Laboratory of Systems Bioengineering, Ministry of Education, Tianjin 300072, China
3 Edinburgh-Tianjin Joint Research Centre for Systems Biology and Synthetic Biology, Tianjin University, Tianjin 300072, China
Dozens of genome-scale metabolic networks have been reconstructed by integrating information from various databases on genes, proteins, metabolites and validated by experiment data from the literature. The reconstructed networks can be used to quantitatively investigate the interactions between components of a biological system at a system level. Such theoretical study could help us understand the organization principle of the large scale network and thus provide guidance to strain optimization through metabolic engineering technology. In this review, we evaluate the methods for the reconstruction, analysis and application of genome-scale metabolic networks. The difficulties and perspectives on this emerging research field are also discussed.
genome-scale, metabolic network, systems biology, metabolic engineering
1995年,第一个物种流感嗜血杆菌Haemophilus influenzae Rd的全基因组序列测序完成[1],随着测序技术的不断发展,测序成本也不断降低,截止至2010年6月4日,已经有1 291个物种全基因组测序完成 (其中细菌1 079株,古细菌83株,真核生物129个[2])。然而如何高效地利用如此庞大的数据成为人们研究的重点,基因组尺度代谢网络就是以基因组序列和注释信息为基础,通过基因-蛋白质-反应相互关系重构模拟生物体的代谢过程。理论上来说,有多少物种的全基因组测序完成,就应该存在多少个对应的基因组尺度代谢网络模型。然而目前只有58个物种的87个基因组尺度代谢网络模型构建完成(GSMNDB:http://synbio.tju.edu.cn/GSMNDB/gsmndb. htm),其数量远远小于已测序物种的数量 (图1)。造成这种情况的原因有很多,其中最主要的3个原因:首先,由于注释算法不完善等因素,基因组中注释出来的基因有很多是未知功能的基因和非编码基因[3];其次,基因组尺度代谢网络构建需要大量的人工校对工作,这个步骤是个非常耗时耗力的工作[4];最后,由于我们对很多物种的生理生化机制了解有限,即使研究最为透彻的大肠杆菌,仍然有很多生命活动的机制都是未知的。
June 9, 2010; Accepted: August 11, 2010
Supported by:National Basic Research Program of China (973 Program) (No. 2007CB707802), National Natural Science Foundation of China (Nos. 20806055, 20875068), Development Project of Science and Technology of Tianjin (No. 05YFGZGX04500), Program of Introducing Talents of Discipline to Universities (No. B06006).
Xueming Zhao. Tel/Fax: +86-22-27406770; E-mail: xmzhao@tju.edu.cn
国家重点基础研究发展计划 (973计划) (No. 2007CB707802),国家自然科学基金 (Nos. 20806055, 20875068),天津市科技发展计划 (No. 05YFGZGX04500),高等学校学科创新引智计划 (No. B06006) 资助。
