对苏丹红GB/T 19681-2005检测方法的探讨与改进
2010-01-25丁轶聪
丁轶聪 王 群 伊 芳
(阜阳市产品质量监督检验所,阜阳 236000)
苏丹红属偶氮系列染料,主要应用于蜡、油彩、汽油等工业领域,是应用广泛的化工合成染色剂。目前已确认了有机偶氮染料的毒性,其中大多数有致癌性,包括苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ和苏丹红Ⅳ[1]。我国目前采用的《食品添加剂使用卫生标准》[2]中允许使用的食品添加剂有1 000多种,但不包括“苏丹红”,即“苏丹红”在食品生产领域是被国家标准明确禁止使用的。由于GB/T 19681-2005国标检测方法[3](高效液相色谱法)发布时间紧促,不可避免地存在一些问题。因此笔者针对国标中存在的一些问题进行探讨,并建立了一种适合大多数检测机构使用的检测灵敏度高、分离效果好的检测方法。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:L-7100型,配有二极管阵列检测器(195~900 nm),日本日立公司;
旋转蒸发器 :R-205型,上海申胜生物科技有限公司;
丙酮、乙腈、正己烷:色谱纯;
甲酸:分析纯;
30%丙酮的正己烷溶液;
中性氧化铝固相萃取柱:2 g/(6 mL),Alumina-N(批号20090815),上海安谱科学仪器有限公司;
苏丹红Ⅰ(≥90.0%)、苏丹红Ⅱ(≥95.0%)、苏丹红Ⅲ(≥96.0%)、苏丹红Ⅳ(≥94.0%)标准物质:德国 Dr.Ehrenstorfer公司。
1.2 色谱分析条件
色谱柱:C18色谱柱(迪马钻石 250 mm×4.6 mm, 5 μm);流动相:0.5%甲酸的乙腈溶液-0.1%甲酸的丙酮溶液(体积比80∶20);流速:1.0 mL/min;柱温:30℃;检测波长:478 nm;进样量:10 μL。
1.3 样品处理
称取0.5 g辣椒油样品于25 mL比色管中,加入9 mL正己烷溶解,难溶解的样品可以适当加温促进溶解。慢慢加入到已用10 mL正己烷过柱的中性氧化铝小柱,待样液完全流出后再加入10 mL正己烷洗柱,弃去全部正己烷淋洗液,用80 mL 30%丙酮的正己烷溶液进行洗脱,收集洗脱液60℃旋蒸浓缩后,用丙酮转移定容至5 mL,经0.45 μm滤膜过滤后待测。
1.4 实验步骤
(1)标准溶液的制备
称取0.01 g左右的苏丹红Ⅰ号、苏丹红Ⅱ号、苏丹红Ⅲ号、苏丹红Ⅳ号标准物质,分别用丙酮溶解并定容至100.0 mL,配制成浓度分别为苏丹红Ⅰ:100 μg/mL、苏丹红Ⅱ:101 μg/mL、苏丹红Ⅲ:108 μg/mL、苏丹红Ⅳ:104 μg/mL的单标,然后分别移取每个单组分标准溶液(以下简称单标)各2.0、5.0、7.0 mL,每相同体积的单标用丙酮合并定容至100.0 mL,得到3种不同浓度的混合标准溶液。
(2)加标回收测定用标准溶液的制备
分别准确移取5.0 mL 1.4(1)配制的单标,用正己烷合并定容至100.0 mL,得到浓度分别为苏丹红Ⅰ:5.4 μg/mL、苏丹红Ⅱ:5.05 μg/mL、苏丹红Ⅲ:5.0 μg/mL、苏丹红Ⅳ:5.2 μg/mL的混合标准溶液。
(3)样品测定
按1.2色谱条件,取检测用3种不同浓度的混合标准溶液和样品处理液各10 μL上机测定,以标样的保留时间定性,以标样和样品的峰高度之比进行定量。
1.5 色谱图
在1.2色谱条件下,对混合标准样品溶液进样分析,色谱图如图1所示。

1—苏丹红Ⅰ; 2—苏丹红Ⅱ; 3—苏丹红Ⅲ; 4—苏丹红Ⅳ图1 标准样品色谱图
2 结果与讨论
2.1 检测波长的选择
国标检测方法选择478 nm检测苏丹红Ⅰ号,出峰后切换成520 nm,检测苏丹红Ⅱ、Ⅲ、Ⅳ号,目前还有很多的基层检测单位使用的液相色谱不具备设置波长梯度检测的功能,根据对标准溶液的光谱扫描,发现4种标准物质在478 nm附近均有较强的吸收,因此实验采用478 nm波长作为检测波长。
2.2 配制标准溶液基质和流动相的选择
按照国标方法用乙醚溶解,用正己烷定容的混合标准溶液在给定的条件下各组分峰分离效果不好,且与样品处理液最终定容基质不同,导致出峰时间不同,峰高有差异[4],改用丙酮溶解、定容,按本实验确定的条件,苏丹红Ⅰ、Ⅱ、Ⅲ、Ⅳ号分离情况较好(见图1)。本方法专门给出了做加标实验的混合标准溶液配制方法,是因为试验中是用丙酮作为解析液,如果再用纯丙酮稀释的混合标准溶液作为加入的标物,在过柱萃取的环节上,标物不能够有效地保留在固相萃取柱上,导致回收率不理想。国标方法在检测过程中使用了梯度洗脱功能,同样不适用于不具备此种功能的液相色谱仪。经试验发现甲酸能够改善峰形,丙酮能够加快出峰时间,使用甲酸乙腈溶液和甲酸丙酮溶液作为流动相,用等度方式同样能得到较好的分离效果,缩短了分析时间,提高了工作效率。
2.3 样液的吸附、解析及浓缩
影响方法回收率的关键环节是样液过固相萃取柱吸附、解析的环节,国标的中性氧化铝活性调整的方法没有明确给出活化后保存方式以及保存期限,实际操作性不强。本实验采用成品SPE小柱,免去了活化和灭活的过程,使用效果较好,但是需要注意的是成品SPE小柱每批次的活性也不尽相同,使用前应采用测定加标回收率的方式加以确认。试验中改用和国标方法不同的30%丙酮的正己烷溶液,增加了洗脱能力,能更彻底地洗脱掉固相萃取柱上的待测物质。
2.4 线性方程、线性范围及检出限
按照1.4(1)配制标准溶液系列,在选定的实验条件下测定并绘制标准曲线。线性方程、线性范围见表1。
对空白溶液进行10次重复测定,以3倍的标准偏差除以工作曲线的斜率,计算苏丹红Ⅰ~Ⅳ号的检出限分别为8、9、9、10 μg/kg。

表1 线性方程、线性范围及检出限
注:y代表含量,x代表响应值。
2.5 精密度试验
取一种浓度的检测用混合标准溶液连续测定6次,测试结果及相对标准偏差见表2。由表2可见方法具有良好的精密度。
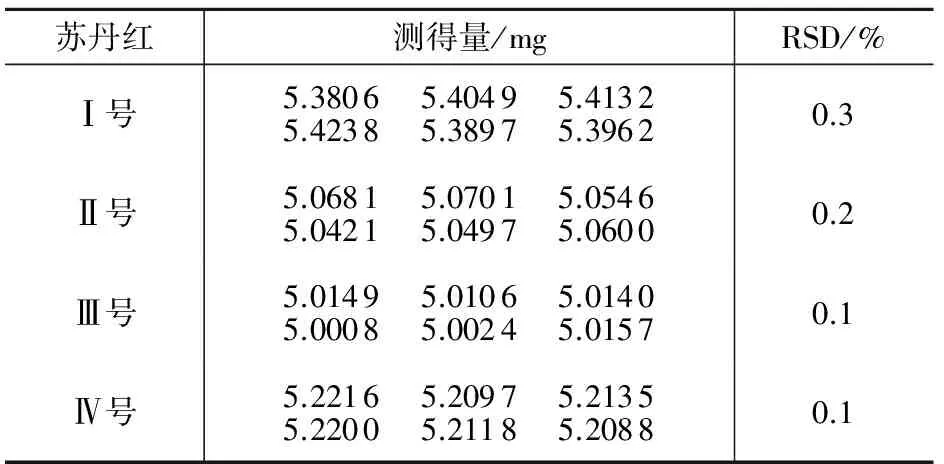
表2 精密度试验
2.6 回收试验
准确移取1.0 mL加标回收测定用标准溶液加入到不含苏丹红的辣椒油样品中,按1.3处理后测定,结果见表3。
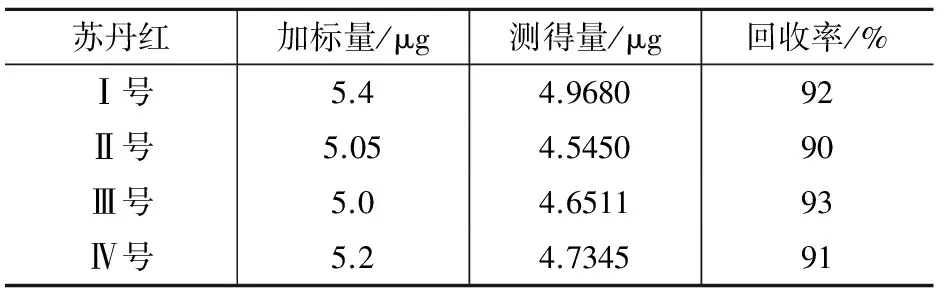
表3 回收试验结果
3 结论
对国标检测方法(高效液相色谱法)测定苏丹红的检测方法进行了改进,改进后的方法具有简便、快捷、准确的特点,方法的线性关系、精密度和回收率均能满足日常检测工作要求,对国标检测方法起到了补充和完善的作用。
[1] 黄晓兰,吴惠勤,黄芳,等. GC-MS/SIM法同时测定食品中的
苏丹红Ⅰ-Ⅳ[J].分析测试学报,2005(4):78-80.
[2] GB 2760-2007 食品添加剂使用卫生标准[S].
[3] GB/T 19681-2005 食品中苏丹红染料的检测方法 高效液相法[S].
[4] 沈兵,奚奇辉,于天祥,等.对食品中苏丹红检测标准的改进[J].理化检验:化学分册,2008,44:535-537
