大前庭水管综合征家系SLC26A4基因传递特征分析△
2010-01-25王艳莉郭玉芬徐百成袁逸铭朱一鸣历建强李倩王秋菊
王艳莉 郭玉芬,2 徐百成 袁逸铭 朱一鸣 历建强 李倩 王秋菊
大前庭水管综合征(large vestibular aqueduct syndrome, LVAS)是一种临床常见的隐性遗传性非综合征型听力障碍性疾病,在儿童和青少年感音神经性聋中占1%~12%[1,2]。它是先天性内耳畸形的一种。临床上将只有前庭水管扩大畸形,不伴有其他内耳发育异常和其他器官系统的异常(如pendred’s Syndrome, Branchiootorenal Syndrome),合并感音神经性听力损失的患者诊断为大前庭水管综合征。其致病基因与Pendred综合征的致病基因相同,即SLC26A4, 定位于人类染色体7q31上,编码Pendrin蛋白[3]。为了明确SLC26A4基因在大前庭水管综合征家系亲子代间的传递规律,阐明家系中患者的发病率及患者后代的发病情况,本研究收集3个大前庭水管综合征家系,对其SLC26A4基因的传递特征进行分析,现报告如下。
1 资料与方法
1.1临床资料的收集 所有家系资料均来自兰州大学第二医院耳鼻咽喉科门诊,该项目的伦理论证由中国人民解放军总医院伦理委员会认可。本研究共收集3个大前庭水管综合征家系,分别命名为502家系、506家系和513家系。影像学检查发现5名大前庭水管综合征患者,包括男性1名,女性4名。
所有纳入者均进行了详细的病史调查和系统的全身体格检查,并根据各成员检测年龄及配合程度分别行纯音测听、听性脑干反应(auditory brainstem response, ABR)及畸变产物耳声发射(distortion product otoacoustic emission, DPOAE)检查。临床病史资料的收集、家系各成员耳部检查及听力学检查由兰州大学第二医院耳鼻喉科专业医师完成。所有听力损失患者均行颞骨轴位高分辨率CT扫描。经各成员本人或其监护人知情同意后抽取外周静脉血5 ml。用Cyrillic2.1软件绘制家系图谱(图1)。对大前庭水管综合征患者进行随访,反复检查其听力。
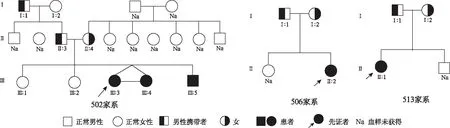
图1 502、506、513家系图谱
1.2听力学检查方法 纯音测听:运用Madsen502便携式听力计,以0.5、1、2、4 kHz的平均听阈作为听力损失的分级标准,参照世界卫生组织(WHO)标准[4]将听力减退划分为5级,即正常≤25 dB HL,轻度:26~40 dB HL,中度:41~60 dB HL;重度:61~80 dB HL;极重度≥81 dB HL。ABR和DPOAE应用美国智听公司Smart EP 听觉诱发电位仪,常规进行测试。
1.3影像学检查 颞骨高分辨率轴位CT示:从半规管总脚到前庭水管外口的1/2处直径大于1.5 mm,即诊断为前庭导水管扩大(EVA)[5]。
1.4基因组DNA提取 应用盐析法在全血中提取基因组DNA,TE溶解,经琼脂糖电泳和紫外分光光度计定量和纯度分析后分装,-20℃保存。
1.5PCR反应 应用在线引物设计软件Primer 3针对外显子8、19和其内含子的侧翼序列设计引物,所有引物由invitrogen公司合成。聚合酶链反应(polymerase chain reaction ,PCR) 采用25 μl反应体系,10×Buffer 2.5 μl,2.5 mM dNTP 2 μl,10 μM引物0.6 μl,Tag酶1.5 U,gDNA 100 ng,加去离子水至25 μl。外显子的引物设计和反应条件参照wang等[6]所述。取2 μl进行琼脂糖凝胶电泳,检测PCR产物。
1.6PCR产物的测序分析 用Millipore纯化板对PCR产物进行纯化,去除多余dNTPs和引物后应用ABI公司 3 730DNA测序仪进行正向和反向直接测序,引物与PCR扩增引物相同。测序结果与来自NCBI上的SLC26A4基因标准序列(NT_007933)在DNAStar 7.0和Sequencher 4.9上进行对比分析,确定突变位点。
1.7家系其他成员的基因检测 针对患者中检测到的SLC26A4基因突变位点,进行对应的序列分析。
2 结果
2.1颞骨高分辨率轴位CT检查结果 颞骨CT共发现5名大前庭水管综合征患者,未合并其他内耳畸形(图2)。
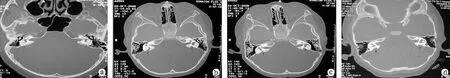
图2颞骨CT图A:正常双侧前庭水管;B:502家系先证者双侧扩大的前庭水管(箭头所示);C: 502家系患者Ⅲ:4双侧扩大的前庭水管(箭头所示);D: 513家系先证者双侧扩大的前庭水管(箭头所示)
2.2临床检查结果 经过系统的全身体格检查,所有纳入者仅表现为听力损失,未见其它系统的异常。3个家系中,父母均正常,患者为同胞,无其他大前庭水管综合征患者。5名大前庭水管综合征患者发病时均不超过3岁,最小3个月,平均1.8岁,只有简单言语发育。
2.3听力学检查结果 所有大前庭水管综合征患者的听力损失程度均达重度或极重度。家系各成员的听力损失情况见表1。
2.4SLC26A4基因检测结果 本研究共发现2种SLC26A4基因的突变类型,即IVS7-2A>G和H723R。5名大前庭水管综合征患者均检测出双等位基因的突变,患者父母为单个突变基因的携带者。家系各成员的基因型见表1。

表1 家系成员听力情况及其SLC26A4基因检测结果
注:*即wild type,野生基因型
3 讨论
本研究对3个大前庭水管综合征家系15名成员的SLC26A4基因进行了分析,发现5名成员携带SLC26A4双等位基因突变,7名成员携带单个等位基因突变,3名正常人。检测到两种SLC26A4基因突变类型,即IVS7-2A>G和H723R,这两种突变类型在国人大前庭水管综合征患者中最常见[6,7]。IVS7-2A>G位于外显子8剪切位点的位置,即内含子7的3’末端,其突变使前体mRNA不能正常剪接,外显子8整个丢失,外显子7和外显子9直接相连,从而导致Pendrin的翻译发生移框或提前终止,影响Pendrin的结构和功能[8]。H723R突变使该位置正常的组氨酸变成精氨酸,影响Pendrin的表达而致病[9]。
在502家系中,先证者Ⅲ:3、双胞胎妹妹Ⅲ:4和其同胞弟弟Ⅲ:5的SLC26A4基因检测结果均为IVS7-2A>G和H723R的复合杂合突变。父母(Ⅱ:3、Ⅱ:4)听力正常,但分别为IVS7-2A>G和H723R的携带者。祖父Ⅰ:1和祖母Ⅰ:2听力正常,但祖父是IVS7-2A>G突变的携带者,祖母未携带致病基因。同胞姐姐Ⅲ:1和Ⅲ:2听力正常,均未携带致病基因。506家系和513家系的构成相似:两家系先证者的基因型均为IVS7-2A>G的纯合突变,父母听力均正常,都为IVS7-2A>G杂合突变携带者,506家系先证者的同胞姐姐和513家系先证者的同胞弟弟听力均正常,血样未获得。由遗传规律可知,子代的染色体分别来自父母双方,分析502家系图谱和各成员的基因型,可以得出如下结论:先证者的祖父携带的IVS7-2A>G突变传递给了先证者的父亲,当他与携带H723R突变的先证者母亲婚配时,两者各自的致病突变分别会遗传给子代,当患者同时遗传了父母双方各自的致病基因,组成自身染色体时就会导致患病。由此可见,SLC26A4突变的等位基因会稳定的由亲代向子代传递,506家系和513家系的SLC26A4基因同样具有这种遗传特征。
大前庭水管综合征属于常染色体隐性遗传病,家系中常仅见一代人发病,先证者的父母为无临床症状的单个突变携带者,患者常携带双等位基因突变。上述3个家系中,均仅见一代人发病,父母听力都正常,但均为致病基因的携带者,其子代中既有大前庭水管综合征患者,又有听力完全正常者,且患者中有男性也有女性,这些特点均符合大前庭水管综合征的遗传特征。根据孟德尔遗传规律,两个携带者婚配后,其子代中患者、携带者和正常者的发生几率分别为25%、50%和25%。由此可以预测,506家系先证者的同胞姐姐和513家系先证者的同胞弟弟为IVS7-2A>G携带者的几率为67%,完全正常的几率为33%[10]。本研究502家系的子代中未发现致病基因的携带者,患者的发病几率是60%(3/5),506家系和513家系中患者的几率均为50%(1/2),均较理论值25%高。那么,先证者父母再生育一个大前庭水管综合征患儿的几率可能会低于25%。因而,临床医生在对大前庭水管综合征家系再生育一个患者的风险进行评估时应谨慎。
大前庭水管综合征的致病基因会由亲代向子代稳定的遗传,上述3个家系中患者的致病基因同样会遗传给其后代,若她(或他)与正常人婚配,其子代均为携带者;若与携带者婚配,子代为携带者或患者,且患者的几率较两个携带者婚配后子代患病的几率增加一倍。因此,倘若婚前进行SLC26A4基因的检测,查明致病突变,并给予积极的遗传指导或进行产前诊断,就可以对大前庭水管综合征聋儿的出生做出早期预警,从而减轻家庭和社会的负担。
近年来,国内学者已在SLC26A4基因热点突变及突变频率、遗传特征等方面进行了比较全面、深入的研究[6,7,11,12]。在此基础上本研究又对3个大前庭水管综合征家系的SLC26A4基因进行序列分析,查明了各家系成员的基因型,明确了各家系的致聋原因,并阐述了该基因由亲代向子代稳定传递的规律,明确了502、506和513家系中子代的患病几率分别为60%、50%和50%,均高于理论值25%;同时对患者与不同类型的配偶婚后所生子代发病情况进行预测、分析,若给予积极的遗传咨询和适当的临床指导,及早进行预防和干预,就可以降低耳聋的发生率,提高患者生活质量。
4 参考文献
1 Griffith AJ, Arts A, Downs C, et al. Familial large vestibular aqueduct syndrome[J].Laryngoscope,1996, 106, 8:960.
2 Mafong DD, Shin EJ,Lalwani AK. Use of laboratory evaluation and radiologic imaging in the diagnostic evaluation of children with sensorineural hearing loss[J].Laryngoscope,2002, 112, 1:1.
3 Abe S, Usami S, Hoover DM., et al. Fluctuating sensorineural hearing loss associated with enlarged vestibular aqueduct maps to 7q31, the region containing the Pendred gene[J].Am J Med Genet,1999, 82: 322.
4 孙喜斌,李兴启,张华.全国第二次残疾人抽样调查听力残疾标准介绍[J].听力学及言语疾病杂志,2006,14:447.
5 Wu CC, Chen YS, Chen PJ, et al. Common clinical features of children with enlarged vestibular aqueduct and Mondini dysplasia[J].Laryngoscope,2005, 115:132.
6 Wang QJ, Zhao YL, Rao SQ, et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China[J].Clin Genet,2007, 72:245.
7 赵亚丽,王秋菊,李庆忠,等.95例前庭水管扩大核心家系SIC26a4基因特异突变图谱[J].听力学及言语疾病杂志,2008, 16:178.
8 Yang JJ, Tsai CC, Hsu HM, et al. Hearing loss associated with enlarged vestibular aqueduct and Mondini dysplasia is caused by splice-site mutation in the PDS gene[J].Hear Res,2005, 199:22.
9 Tsukamoto K, Suzuki H, Harada D, et al. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: A unique spectrum of mutations in Japanese[J].Eur J Hum Genet,2003, 11: 916.
10 赵亚丽,王秋菊,兰兰,等, 大前庭水管综合征家系SLC26A4基因突变分析[J].中华耳科学杂志,2006, 4:322.
11 李琦,戴朴, 黄德亮,等.SLC26A4基因IVS7-2A>G突变在中国不同地区和民族重度感音聋患者中分布频率的观察[J].中华耳鼻咽喉头颈外科杂志,2007, 42:5.
12 Dai P, Li Q, Huang D, et al. SLC26A4 c.919-2A>G varies among Chinese ethnic groups as a cause of hearing loss[J].Genet Med,2008,10:586.
