青霉素扩环半合成头孢菌素研究进展
2010-01-10郭晓强张信中
郭晓强,甘 亚,张信中,颜 军,姚 倩
(1.成都大学生物产业学院,四川成都 610106; 2.中国医药集团四川抗菌素工业研究所有限公司,四川成都 610051)
0 引 言
自1929年,Fleming偶然发现青霉素以来,科学家们为了克服青霉素抗菌谱较窄、易引起过敏性休克和性质不稳定等缺点,在继续对青霉素进行研究的同时不断努力寻找更好的抗生素.1945年,Brotzn在对意大利的Sardinia近海污水进行药物研究时发现了一种对革兰氏阳性细菌(G+)和革兰氏阴性细菌(G-)有强抑制的头孢菌素(Cephalosporin).头孢菌素的发现和使用对人类抗感染治疗具有重要意义.但尽管人们经过大量的努力,头孢菌素仍然难以达到青霉素的发酵水平,且面临头孢菌素发酵产物混合物分离和纯化难度大等诸多问题.1963年,Morin等首先将青霉素G或青霉素V氧化,扩环为3-去乙酰氧基头孢菌素,再以五氧化二磷去侧链,转化为7-氨基脱乙酰氧基头孢烷酸(7-ADCA),从而证明由青霉素母核扩环制备头孢母核的可行性,此方法为廉价的青霉素转化为较高附加值的头孢菌素提供了可能[1,2].从此,半合成头孢菌素产量以及品种不断增加,目前已发展到第四代头孢菌素共计50余种,其品种数量居各类抗生素首位[3].
1 头孢菌素主要合成方法
1.1 头孢菌素的全合成
1966年,Woodward首次在实验室全合成了头孢菌素C和先锋霉素I号[4],而目前能用全合成方法工业化生产的仅限于1-碳头孢菌素(1-carbacephalosporin)类的氯碳头孢菌素等少数几个品种[5,6],但因其产量较低而不适合于大规模生产.
1.2 头孢菌素半合成[6]
1.2.1 以7-ACA为母核半合成头孢菌素.
7-ACA主要是由头孢菌素C(CPC)经化学法或酶法裂解得到,可对其进行相关结构改造制备各类头孢菌素.同时,由于其市场价格相对便宜等原因, 7-ACA仍然是半合成头孢菌素的主要中间体.
1.2.2 青霉素扩环半合成头孢菌素.
20世纪70年代,随着β-内酰胺抗生素作用机理和构效关系研究的显著进步,研究人员开始探索β-内酰胺母核修饰的可能性,并开始研究利用经济的青霉素G为原料经扩环制备而得头孢菌素中间体,如,7-ADCA、GCLE(GCLH)、3-羟基头孢烯、3-环外亚甲基头孢烷酸和丙二烯羧酸酯类氮杂环丁酮等,并以此来半合成头孢菌素类、1-氧头孢菌素类和头霉素类等新型抗生素.
2 青霉素扩环制备C3-位功能化头孢菌素中间体合成及应用
在对头孢菌素半合成过程中除了C7-位的修饰以外,C3-位取代基改变及功能化将影响药物动力学性质、药物溶解性和生物活性等,因此对C3-位改造成为头孢菌素半合成的重要途径[3].
2.1 7-ADCA的合成及应用
目前,采用青霉素工业盐扩环转化成7-苯乙酰胺基去乙酰氧基头孢烷酸(7-ADCA)的工艺已经很成熟,大部分的医药企业都采用此路线合成 7-ADCA,该合成的关键点是酶法裂解侧链.通过7-ADCA可以合成当前全球半合成抗生素销售前列的头孢氨苄(cefalexin)、头孢拉定(cefradine)、头孢羟氨苄(cefadroxil)等产品(见图1).
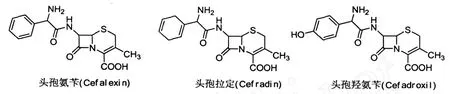
图1 以7-ADCA为原料半合成的主要品种
2.2 GCLE和GCLH的合成及应用
7-苯乙酰胺-3-氯甲基头孢烷烯酸对甲氧基苄酯(p-methoxybenzyl-7-Phenyl-acetamido-3-chloromethyl-3-cephem-4-carboxylate,GCLE)和7-苯乙酰胺-3-氯甲基头孢烷烯酸二苯甲酯(diphenylmethyl-7-phenylacetamido-3-chloromethyl-3-cephem-4-carboxylate,GCLH),是从青霉素通过化学转化合成3’-取代的头孢菌素类抗生素的关键中间体[7],其合成路线如图2所示.
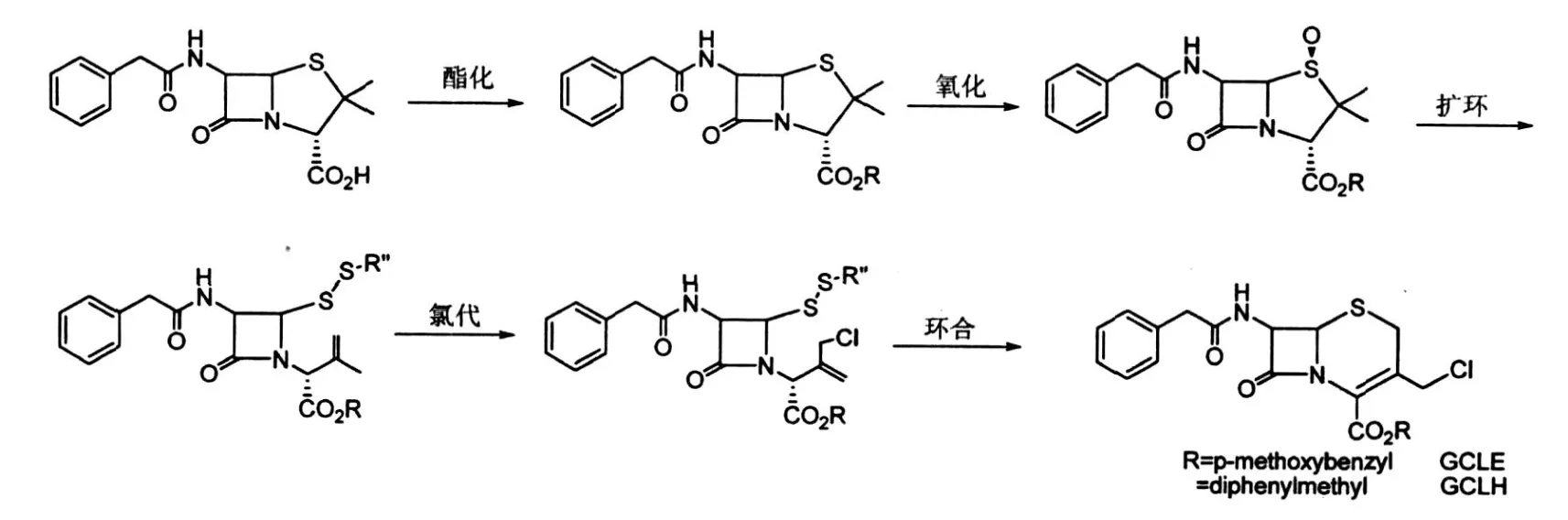
图2 GCLE、GCLH的合成路线
由于GCLE和GCLH分子中C3-位氯甲基的高活性为合成C3-位含有不同基团取代的头孢菌素类抗生素提供了简捷高效的合成方法.根据药物设计基本原理,GCLE(GCLH)可合成得到:①C3-位含双键的头孢菌素类抗生素,如头孢克肟(cefixime)、头孢托仑(cefditoren)等;②C3-位含硫甲基的头孢菌素类抗生素,如头孢唑啉(cefazoline)、头孢曲嗪(cefatrizine)等;③C3-位含氮甲基的头孢菌素类抗生素,如头孢他啶(ceftazidime)、头孢匹罗(cefpirome)等.其合成产品包括了大多数第一代头孢菌素类抗生素到第四代头孢菌素类抗生素.图3显示了以GCLE和GCLH为母核半合成的主要品种.
2.3 3-羟基头孢烯和3-环外亚甲基头孢烷酸的合成及应用
3-羟基头孢烯是制备一系列在Δ3-头孢烯骨架C3-位直接相连氢或杂原子如氯、甲氧基的口服头孢菌素抗生素的通用中间体.其合成可以通过对头孢菌素衍生物C3-位的乙酰氧基进行修饰制得[5,10].从经济角度考虑,由廉价的青霉素G出发,扩环转化为3-羟基头孢烯是一条更适合工业化的合成路线(见图4).
早期,研究人员通过氧化环外双键而得到3-羟基头孢烯[11],而Hamashima等采用先氧化后环合的合成策略,并且使用“一锅烩”的工艺,制备的烯醇收率达到70%.由于该工艺使用臭氧氧化,在生产过程中存在一定的危险性,后来新的氧化试剂RuCl3/ HIO4/CuSO4已经代替臭氧用于氧化双键[13],从而使烯醇的收率提高到80%.此外,使用BiCl3/Sn或TiCl4/Sn做还原性环合反应得到目标产物,烯醇收率可达85%.目前上市的产品有头孢唑肟(ceftizoxime)、头孢布坦(ceftibuten)、头孢沙定(cefroxadine)、头孢克罗(cefaclor)等.图5显示了以3-羟基头孢烯和3-环外亚甲基头孢烷酸为母核半合成的主要品种.
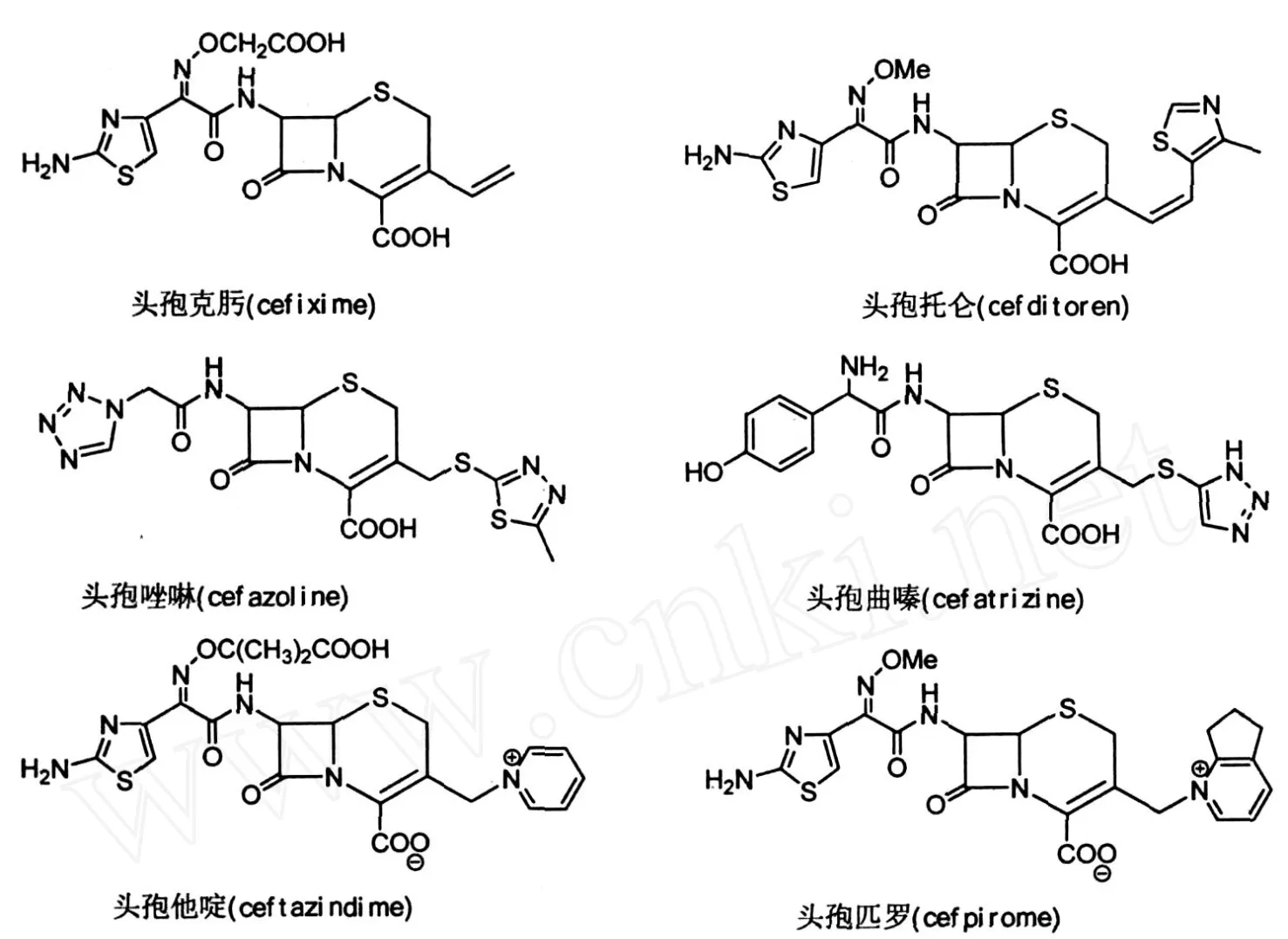
图3 以GCLE和GCLH为母核半合成的主要品种

图4 3-羟基头孢烯和3-环外亚甲基头孢烷酸合成路线
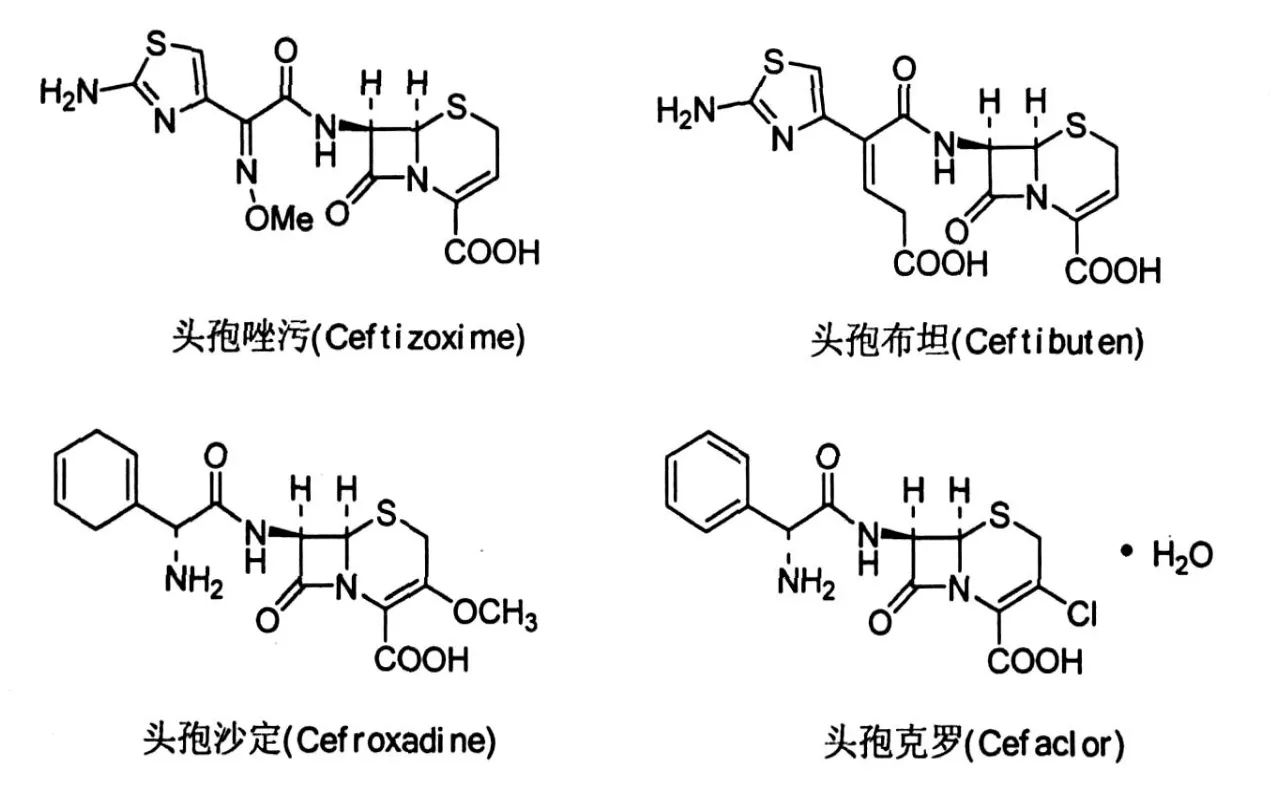
图5 以3-羟基头孢烯和3-环外亚甲基头孢烷酸为母核半合成的主要品种
2.4 丙二烯羧酸酯类氮杂环丁酮的合成及应用
丙二烯羧酸酯类氮杂环丁酮中间体是合成3-位直接与氮、硫、氧、氯等杂原子相连的头孢菌素中间体,同时其也是合成3-羟基头孢烯和3-环外亚甲基头孢烷酸两种化合物的重要原料[14-16].Tanaka等开展了大量的以丙二烯羧酸酯类氮杂环丁酮为中间体的头孢菌素半合成工作,特别是合成3-正头孢菌素化合物[17].图6为丙二烯羧酸酯类氮杂环丁酮的合成路线.

图6 丙二烯羧酸酯类氮杂环丁酮的合成路线
3 青霉素扩环合成头霉素
1971年,研究人员首先从某些链霉菌中发现头霉素(Cephamycins)类抗生素,其特征是在头孢菌素的C7-位上具有α-甲氧基,增强了抗革兰阴性菌的作用,并且对β-内酰胺酶的稳定性优于多数头孢菌素.这些物质的发现开创了对新型化合物7α-甲氧基头孢菌素的合成研究工作:氧化侧链酰胺的氮,随后经消除反应形成亚胺而把氧化点转移到7-位碳原子上,其后立体选择性地在C7-位引入α-甲氧基[18-20].图7为青霉素扩环合成头霉素工艺路线及品种.

图7 青霉素扩环合成头霉素工艺路线及品种
4 青霉素扩环合成1-氧头孢菌素
1978年,Suarato等[21]从青霉素亚砜酯出发,经一个新颖的开环方法制得光学活性的4-酰氧基氮杂环丁烷酮,从而启发人们以6-差向青霉素为起始物合成7-差向-1-氧头孢菌素.Shionogi公司的科研人员在氧头孢菌素的合成研究方面做出了重要贡献,他们提出了许多有用的合成策略[22].目前,已经有氟氧头孢菌素(flomoxef)和拉氧头孢菌素(latamoxef)两个产品上市.图8为青霉素扩环合成1-氧头孢菌素(1-Oxacephems)工艺路线及品种.
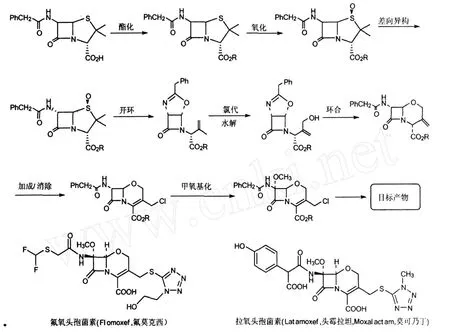
图8 以青霉素扩环合成1-氧头孢菌素的工艺路线与品种
5 结 语
由于现代发酵技术降低了青霉素的生产成本,通过将廉价的青霉素G经扩环制备而得各种头孢菌素中间体及其产品符合我国国情.同时,通过青霉素转化为头孢菌素的合成路线对消化我国过剩的青霉素产品和提高相关产品附加值将起到积极的促进作用.但由于我国相关医药企业缺乏技术创新意识,只停留在上游产品的低水平重复建设上,技术含量低、附加值低的产品过剩,而技术含量高、附加值高的品种不足,真正意义上的新化学药物研制和开发尚末形成气候.基于此,本文认为在采用青霉素扩环半合成头孢菌素工艺路线时应注意以下两点:
(1)突破青霉素G转化的合成工艺技术难点,运用新理论、新技术研究和改进工艺,提高工艺过程的技术创新性,从而增加产品的附加值.同时,关注发达国家即将失去专利保护的优秀药物品种,以此来开发出一系列我国需求量大的品种.
(2)紧跟国外新品种的开发动向,进行新衍生物的开发,并积极从文献上寻找已报道的优秀的β-内酰胺类抗生素的侧链,将此侧链引入新化合物中,走高起点、快发展的道路.
[1]Morin R B,Jackson B G,Mueller R A,et al.Chemistry of Cephalosporin Antibiotics.III.Chemical Correlation of Penicillin and Cephalosporin Antibiotics[J].J Am Chem Soc,1963,85 (12):1896-1897.
[2]Chauvette R R,Pennington P A,Ryan C W,et al.Chemistry of Cephalosporin Antibiotics.XXI.Conversion of Penicillins to Cephalexin[J].J Org Chem,1971,36(9):1259-1267.
[3]刘姝晶,陈耀祖.头孢菌素C-3位功能化及合成中间体的研究进展[J].国外医药抗生素分册,1999,20(6):241-245.
[4]Woodward R B,Heusler K,G osteli J,et al.The Total Synthesis of Cephalosporin C[J].J Am Chem Soc,1966,88(4):852-853.
[5]Farina V,Baker S R,Hauck S I.A General Route to3-functionalized3-norcephalosporins[J].J Org Chem,1989,54(20):4962 -4966.
[6]Bodurow C C,Boyer B D,Brennan J,et al.An Enantioselective Synthesis of Loracarbef(LY163892/KT3777)[J].Tetrahedron Letters,1989,30(18):2321-2324.
[7]刘家健.头孢菌素类品种研发与生产现状探讨[J].中国抗生素杂志,2006,31(2):100-106.
[8]王文梅.头孢菌素中间体GCLE的合成及应用[J].精细与专用化学品,2003,11(10):19-20.
[9]杨艺虹,张 珩,杨建设.头孢菌素药物中间体 GCLE和GCLH的合成技术[J].化工科技市场,2004,27(5):19-22.
[10]Robert R.Chauvette R R,Pennington P A,et al.Chemistry of Cephalosporin Antibiotics.XXIX.3-Halo-and3-methoxy-3-cephems[J].J Am Chem Soc,1974,96(15):4986-4987.
[11]Kukolja S.Recent Advances in the Chemistry ofβ-Lactam Antibiotics[M].London:The Chemical Society Burlington House, 1977:181-188.
[12]Hamashima Y.Recent Advances in the Chemistry ofβ-Lactam Antibiotics[M].London:The Chemical Society Burlington House,1977:243-251.
[13]Tanaka H,Taniguchi M,Kameyama Y,et al.A Facile Access to 3-Hydroxycephems from Penicillin G through Bi/Sn or Ti/Sn Redox-Promoted Cyclization of4-(Phenylsulfonylthio)azetidinones[J].Chemistry Letters,1990,19(10):1867-1868.
[14]Taniguchi M.Development and Industrialization of New Intermediate3-chloromethyl-Δ3-cephems for Cephaiosporin Antibiotic Synthesis[J].Nippon Kagaku Kaishi,1995(8):577-587.
[15]Tanaka H,Kamayama Y,Sumida S,et al.A New Short Cut Route to3-Norcephalosporins[J].Synlett,1991(12):888-890.
[16]T orii S,Tanaka H,Sasaoka M,et al.Process for Preparing an Allenyl Beta-lactam Compound[P].US,5986091.1999-11-16.
[17]Tanaka H,Yamaguchi Y,Sumida S,et al.Generation and Reaction of Copper(I)Hydride in the Copper(I)Chloride Tributyltin Hydride-NMP System:Synthesis of3-norcephalosporin [J].J Chem Soc,1999,8(1):3463-3468.
[18]Baldwin J E,Urban FJ,Cooper R D G,et al.Direct6-methoxylation of Penicillin Derivatives.Convenient Pathway to Substituted Beta-lactam Antibiotics[J].J Am Chem Soc,1973,95 (7):2401-2403.
[19]K oppel GA,K oehler R A.Functionalization of C6(7)of penicillins and cephalosporins.One-step stereoselective synthesis of7-alpha-methoxycephalosporin C[J].J Am Chem Soc,1973,95 (7):2403-2404.
[20]Raymond A.Firestone R A,Chistensen B G.Functionalization of penicillins at C-6via N-acylimines forAbstracting.6-Hydroxy penicillin.Substituted Penicillins and Cephalosporns.VIII[J].J Org Chem,1973,38(7):1436-1437.
[21]Suarato A,Galliani C.A New Route to Optically Active4-acyloxy azetidin-2-ones[J].Tetrahedron Letters,1978,19(42): 4059-4062.
[22]Y oshioka M,Tsuji T,Uyeo S,et al.Stereocontrolled StraightforwardSynthesis of3-substituted methyl7α-methoxy-1-oxacephems[J].Tetrahedron Letters,1980,21(4):351-354.
