1-(2-羟乙基)-2-烷基咪唑啉缓蚀剂在铁表面吸附的分子动力学模拟
2010-01-04姜娟娟任振甲于维钊于立军乔贵民
张 军,姜娟娟,任振甲,于维钊,于立军,乔贵民
(1.中国石油大学 物理科学与技术学院,山东东营 257061;2.海洋石油工程(青岛)有限公司,山东青岛 266520;3.中国石油大学石油工程学院,山东青岛 266555)
1-(2-羟乙基)-2-烷基咪唑啉缓蚀剂在铁表面吸附的分子动力学模拟
张 军1,姜娟娟2,任振甲1,于维钊3,于立军1,乔贵民1
(1.中国石油大学 物理科学与技术学院,山东东营 257061;2.海洋石油工程(青岛)有限公司,山东青岛 266520;3.中国石油大学石油工程学院,山东青岛 266555)
采用分子动力学(MD)模拟方法,分析液相条件下6种不同烷基链长的1-(2-羟乙基)-2-烷基咪唑啉缓蚀剂在Fe(001)表面的吸附。结果表明:6种缓蚀剂分子的极性头基均会吸附在铁表面,且头基中的咪唑环在铁表面上近似平行吸附,而分子的烷基碳链则背离金属表面,并以一定的倾角指向液相;随分子烷基链长的增加,缓蚀剂与铁表面的结合强度逐渐增大,同时缓蚀剂膜的致密性也逐渐增大;模拟的6种缓蚀剂分子缓蚀性能与实验结果吻合。
吸附;分子动力学;模拟;咪唑啉缓蚀剂;铁表面
多数有机缓蚀剂[1]为吸附型缓蚀剂,在金属表面吸附时会形成保护膜,可阻止腐蚀介质与金属表面的接触,从而达到减缓金属腐蚀的目的。缓蚀剂分子的物理、化学性质在阻碍金属腐蚀上起着重要的作用,虽然各种实验和理论方法被用来研究缓蚀剂分子的结构性质,但是对缓蚀剂分子与金属表面之间的相互作用了解甚少[2]。分子动力学(molecular dynamics,MD)模拟方法能够提供有关缓蚀剂分子在金属表面吸附过程的详细信息,为深层次探讨缓蚀机制创造了条件,目前已发展成为一种可在分子水平对复杂体系进行研究的有效手段。采用MD方法研究缓蚀剂在金属表面吸附已取得一些有价值的研究成果[1-5]。1-(2-羟乙基)-2-烷基-咪唑啉是一类优良的缓蚀剂,可有效抑制 H2S对碳钢的腐蚀[6]。笔者采用MD模拟方法考察6种不同烷基链长的 1-(2-羟乙基)-2-烷基-咪唑啉缓蚀剂在铁表面的吸附,并对其吸附行为的微观机制进行分析,在计算结果和分析讨论的基础上,将6种缓蚀剂缓蚀性能的理论评价结论与实验结果进行比较。
1 计算方法
采用Accelrys公司的计算软件Material Studio。力场模型选用出自量子力学从头算的Compass力场[7],利用Visualizer模块构建铁晶体、缓蚀剂分子及水分子,能量优化[8]及MD模拟均通过Discover模块完成。模拟温度为298 K,采用Andersen恒温器进行控制[9],各分子的起始速度由 Maxwell-Boltzmann[10]随机分布产生。利用周期性边界条件和时间平均等效于系综平均等基本假设,运用Velocity Verlet算法求解牛顿运动方程。范德华和库仑相互作用采用Group Based[11]算法,截断半径取1.5 nm,截断距离之外的分子间相互作用按平均密度近似方法进行校正,模拟立方体边长大于2倍的截断半径[12]。6种咪唑啉类缓蚀剂的分子结构和缓蚀效率见表1。

表1 6种咪唑啉类缓蚀剂的分子结构和缓蚀效率Table 1 Conformations and experimental inhibition efficiencies for six imidazoline inhibitors
以D分子为例,分子与铁表面相互作用的计算模型如图1所示。选取Fe(001)晶面为吸附表面,表面体系为3.1530 nm×3.1530 nm×1.5765 nm,厚度为12层共计1 452个铁原子,模拟过程中“冻结”铁表面体系中的所有原子。中间层分为2类,均利用Amorphous Cell模块构建完成,一类是包含1 000个H2O分子和1个缓蚀剂分子的体系(密度为1.0 g/cm3),另一类是包含1 000个H2O分子和9个缓蚀剂分子的体系(密度约为0.96 g/cm3)[13]。最上层水体系的构建也通过Amorphous Cell模块实现,其包含500个水分子(密度为1.0 g/cm3),模拟过程中“冻结”该层所有的原子坐标,使其形成“一面墙”,可有效抑制中间层的分子向上扩散,避免因周期性结构造成晶体界面底层原子与溶剂层原子的重复计算。
缓蚀剂在铁表面吸附行为的模拟计算通过Discover模块中的NVT系综实现。对于图1(a)体系,模拟时间为500 ps,步长为1.0 fs,每1000步输出1帧,通过分析体系温度和能量随时间演化曲线可知,300 ps后,温度波动在(298±10)K,能量偏差在0.5%左右,表明体系已达到平衡,故选择后100 ps进行取样平均,求得各参量的统计平均值。对于图1(b)体系,由于缓蚀剂分子间存在相互作用,体系达到平衡所需时间较长,因此选取模拟时间为1000 ps,步长为1.0 fs,每1000步输出1帧,同样选择后100 ps进行取样平均,求得各参量的统计平均值[14]。
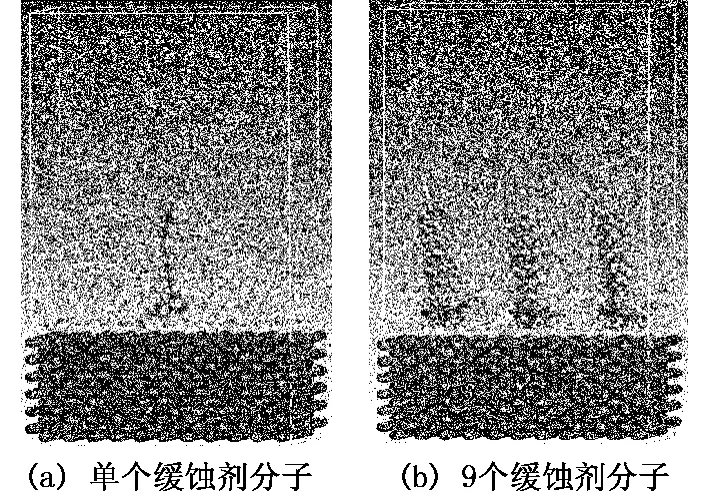
图1 液相条件下D缓蚀剂分子在Fe(001)表面的初始吸附构型Fig.1 Initial configurations of inhibitor molecule D adsorbed on Fe(001)surface in liquid phase
2 结果分析
2.1 缓蚀剂分子在铁表面的平衡吸附构型
图2为液相条件下6种缓蚀剂分子在铁表面的平衡吸附构型。从图2可以看出,与初始构形(图1)相比,平衡时缓蚀剂分子的咪唑环倾向于平行吸附在铁表面上,且烷基长链发生弯曲形变并以一定的倾角指向溶液。这种吸附方式既有利于缓蚀剂分子的头基(咪唑环和羟乙基)在铁表面形成稳定的吸附、改变金属表面的界面状态、提高腐蚀反应的活化能,又有利于烷基碳链在金属表面形成疏水性的保护膜、阻碍腐蚀介质向金属表面的转移,从而达到减缓或抑制腐蚀的目的。

图2 缓蚀剂单分子在铁表面吸附的平衡构型图Fig.2 Equilibrium adsorption configurations on Fe(001)surface for single molecule
表2中给出咪唑环的取向角(β)、咪唑环质心到铁表面的距离(d)及烷基长链在铁表面上的吸附角(θ)的统计平均值。由表2可知:6种缓蚀剂分子咪唑环取向角的统计平均值均小于14.27°,表明咪唑环具有平行吸附的趋势,这与杨怀玉等人的研究结论[15]是一致的,这是由于咪唑环上的N(4),N(7)和C(8)原子具有较大的电负性,可提供孤对电子与铁表面形成配位结合[16],从而使咪唑环有平行吸附于铁表面的趋势;咪唑环取向角介于10.59°~14.27°,且咪唑环质心与铁表面的距离介于0.288~0.299 nm,这充分表明咪唑环的平行吸附构型受烷基链长的影响比较小;随烷基链长增加,烷基链的吸附角逐渐增大,即碳链向金属表面倾倒的趋势随链长增加而增大,这是由于随链长增加,烷基链与铁表面原子之间的范德华作用增强造成的。
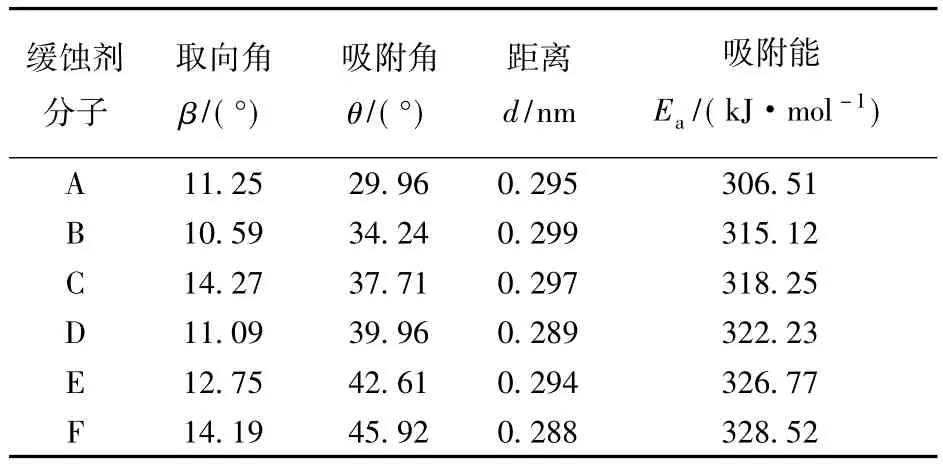
表2 咪唑啉分子在铁表面的取向角、吸附角、距离和吸附能的统计平均值Table 2 Statistic average value of orientation angle,adsorption angle,distances and adsorption energy on iron surface for imidazoline molecules
对比真空条件下咪唑啉分子吸附构型(头基和烷基长链均平行吸附在 Fe(001)表面)[16],在液相条件下缓蚀剂分子的吸附形态发生了明显变化,这与溶剂水分子的影响密切相关。由于溶剂化效应,烷基长链将发生扭曲变形,且水分子在铁表面附近的堆积阻碍了烷基长链向金属表面的倾倒。因此,吸附达到平衡时,缓蚀剂分子的烷基长链将与铁表面形成一定的夹角,而头基因具有较强的活性,可驱替铁表面附近的水分子从而平行吸附于铁表面。
2.2 缓蚀剂分子与铁表面的相互作用
液相条件下,缓蚀剂分子A~F在铁表面的平衡吸附构型的差异将直接影响缓蚀剂分子与金属表面的结合方式及结合强度。为进一步描述缓蚀剂与铁表面的相互作用,计算了A~F在Fe(001)表面的吸附能(Ea)。吸附能可以反映缓蚀剂分子与金属表面的结合强度,其数值越大,表明缓蚀剂分子在金属表面的吸附越稳定,缓蚀性能就越好。缓蚀剂分子在铁表面的吸附能由如下公式计算[17]:
Ea=(EFe+Ei)-EFe+i. (1)式中,EFe为未吸附分子时金属表面体系的能量,kJ/mol;Ei是缓蚀剂分子的能量,kJ/mol;EFe+i是吸附的缓蚀剂分子和金属表面体系的总能量,kJ/mol。
缓蚀剂单分子A~F在Fe(001)表面的吸附能数据见表2。从表中可以看出:随烷基链长的增加,缓蚀剂分子的吸附能逐渐增大,这是由于缓蚀剂分子主要通过极性头基吸附在金属表面,烷基链越长,分子的“柔性”越强,缓蚀剂分子越容易通过烷基链的形变来获取稳定的吸附构型(即体系能量比较低);烷基链越长,吸附角越大,烷基链越易贴近金属表面,增加了分子与金属表面的作用区域,因此吸附能数值随烷基链增加而增大。缓蚀剂的吸附能越大,表明缓蚀剂与金属表面的结合越牢固,其缓蚀性能就越好,故6种咪唑啉缓蚀剂分子缓蚀性能由低到高依次为A,B,C,D,E,F,这与实验方法测得的缓蚀效率的变化规律相吻合[6]。水分子在Fe(001)表面的吸附能[16]为23.40 kJ/mol,A ~F分子在铁表面的吸附能均明显大于水分子的,说明缓蚀剂分子A~F均可驱替水分子而吸附在金属表面上。缓蚀剂分子在铁表面的稳定吸附,有利于缓蚀剂分子在金属表面形成一层保护膜,可有效降低腐蚀介质与金属表面的接触,从而抑制金属表面的腐蚀。
2.3 缓蚀剂膜在Fe(001)表面的吸附
为细致描述分子的吸附成膜,考察了多个缓蚀剂在Fe(001)表面的吸附行为。图3为缓蚀剂分子A~F在Fe(001)表面成膜的平衡构型。可以看出:缓蚀剂分子的极性头基吸附于金属表面,而烷基链相互交织形成一层疏水膜,覆盖在金属表面;随烷基链长的增加,链与链之间的交织越来越明显,缓蚀剂膜在铁表面的覆盖程度也随之提高。这主要是由于随链长的增加,烷基链的柔性增强,烷基链通过交织形成疏水膜的致密性也相应提高[18],可有效抑制水分子向金属表面扩散,进而提高缓蚀剂的缓蚀性能。

图3 缓蚀剂在铁表面吸附成膜的平衡构型Fig.3 Equilibrium configurations of inhibitor monolayer adsorbed on Fe surface
为描述缓蚀剂膜阻碍水分子扩散的行为,以分子F为例,分析了在有、无缓蚀剂条件下水分子的浓度分布随距铁表面高度的变化规律[19]。图4为水分子的数密度分布曲线。可以看出:添加缓蚀剂前后,水分子数密度分布曲线的第一个峰值均出现在距铁表面高度0.2600 nm处;与无缓蚀剂时相比,添加缓蚀剂后峰值由6.14减小至2.07,这是由于缓蚀剂分子的头基驱替铁表面附近的水分子造成的;在距铁表面高度为0.411~1.740 nm区间内,添加缓蚀剂后水分子的数密度均小于无缓蚀剂的情况,这是因为烷基长链在远离金属表面附近通过交织作用形成一层疏水膜,从而排挤该区域的水分子远离铁表面;在距铁表面高度为1.740~2.950 nm区间内,添加缓蚀剂后水分子数密度均大于无缓蚀剂时,这是由于0~1.740 nm区间内被驱替的水分子迁移至该区域所致。
由此可知,在咪唑啉分子头基相同的情况下,分子的烷基链长是影响其缓蚀性能的主要因素,随烷基链长的增加,缓蚀剂分子不仅与金属基体结合更牢固,而且缓蚀剂膜自身的致密性和对金属表面的覆盖度也随之增加,有利于缓蚀性能的提高。
图4 有、无缓蚀剂条件下金属表面水分子的数密度分布
Fig.4 Number density profiles for water molecules on metal surfaces with and without inhibitors
3 结束语
采用分子动力学模拟方法,研究液相条件下6种 1-(2-羟乙基)-2-烷基咪唑啉缓蚀剂在 Fe(001)表面的吸附行为,并分析烷基链长对缓蚀剂吸附行为及其缓蚀性能的影响规律。在液相条件下,缓蚀剂分子A~F的咪唑环均平行吸附于铁表面,其吸附形态受烷基链长的影响比较小,烷基长链则背离金属表面并以一定的倾角指向液相,并发生不同程度的扭转形变。随烷基链长的增加,缓蚀剂分子与铁表面的结合逐渐增强,同时烷基链交织形成疏水膜的致密性也随之增加,因而具有更好的缓蚀性能。6种缓蚀剂分子缓蚀性能的理论评价结果由低到高依次为A,B,C,D,E,F,与文献中缓蚀效率的变化规律完全吻合。
[1] XIA Shu-wei,QIU Meng,YU Liang-min,et al.Molecular dynamics and density functional theory study on relationship between structure of imidazoline derivatives and inhibition performance[J].Corrosion Science,2008,50(7):2021-2029.
[2] ZHANG Jun-ping,ZHANG Qiu-yu,REN Hua,et al.Inhibition performance of 2-mercaptobenzothiazole derivatives in CO2saturated solution and its adsorption behavior at Fe surface[J].Applied Surface Science,2007,253(18):7416-7422.
[3] ANDREAS K,SELMA H.Molecular dynamics simulations of the adsorption of industrial relevant silane molecules at a zinc oxide surface[J].Journal of Chemical Physics,2003,119(18):9719-9728.
[4] ANDREAS K,SAMUEL A F,ALEXEY A S,et al.In-teraction of adsorbed organosilanes with polar zinc oxide surfaces:a molecular dynamics study comparing two models for the metal oxide surface[J].Chemical Physics Letters,2004,393(1/3):107-111.
[5] ANDREAS K,GERHARD E N.Adsorption of organosilanes at a Zn-terminated ZnO(0001)surface:molecular dynamics study[J].Langmuir,2006,22(19):8036-8042.
[6] LUIS S Z R,ESTRADA A,BENAVIDES A,et al.Control de la corrosion de acero al carbon en ambientes de acido sulfhldrico por 1-(2-Hidroxietil)-2-alquil-imidazolinas y sus correspondientes precursores amldicos[J].J Mex Chem Soc,2002,46(4):335-340.
[7] SUN H,REN P,FRIED J R.The compass force field:parameterization and validation for phosphazenes[J].Computational and Theoretical Polymer Science,1998,8(1/2):229-246.
[8] ZHANG Xue-fen,LU Gui-wu,WEN Xiao-ming,et al.Molecular dynamics investigation into the adsorption of oil—water—surfactant mixture on quartz[J].Applied Surface Science,2009,255(13/14):6493-6498.
[9] ANDREA T A,SWOPE W C,ANDERSEN H C.The role of long ranged forces in determining the structure and properties of liquid water[J].J Chem Phys,1983,79(4):4576-4584.
[10] AHUNBAY M G,KARVAN O,ERDEM-ŞENATALAR A.Mtbe adsorption and diffusion in silicalite-1[J].Microporous and Mesoporous Materials,2008,115(1/2):93-97.
[11] PRATHAB B,SUBRAMANIAN V,AMINABHAVI T M.Computation of surface energy and surface segregation phenomena ofperfluorinated copolymers and blends:a molecular modeling approach[J].Polymer,2007,48(1):417-424.
[12] ANDERSEN H C.Molecular dynamics simulations at constant pressure and/or temperature[J].J Chem Phys,1980,72(4):2384-2393.
[13] WU Chao-fu,XU Wei-jian.Atomistic simulation study of absorbed water influence on structure and properties of crosslinked epoxy resin[J].Polymer,2007,48(18):5440-5448.
[14] 刘林法,刘金祥,张军,等.缓蚀剂膜抑制腐蚀介质扩散行为的分子动力学模拟[J].高等学校化学学报,2010,31(3):537-541.
LIU Lin-fa,LIU Jin-xiang,ZHANG Jun,et al.Molecular dynamics simulation of the corrosive medium diffusion behavior inhibited by the corrosion inhibitor membranes[J].Chemical Journal of Chinese Universities,2010,31(3):537-541.
[15] 杨怀玉,陈家坚,曹楚南,等.H2S水溶液中的腐蚀与缓蚀作用机制的研究[J].中国腐蚀与防护学报,2003,23(2):75-78.
YANG Huai-yu,CHEN Jia-jian,CAO Chu-nan,et al.Study on corrosion and inhibition mechanism in H2S aqueous solutions[J].Journal of Chinese Society for Corrosion and Protection,2003,23(2):75-78.
[16] 张军,胡松青,王勇,等.1-(2-羟乙基)-2-烷基-咪唑啉缓蚀剂缓蚀机制的理论研究[J].化学学报,2008,24(7):1239-1244.
ZHANG Jun,HU Song-qing,WANG Yong,et al.Theoretical investigation on inhibition mechanism of 1-(2-Hydroxyethyl)-2-alkyl-imidazoline corrosion inhibitors[J].Acta Chimica Sinica,2008,24(7):1239-1244.
[17] LI Yu-qi,LI Xin,LI Ying,et al.Selective recognition of veterinary drugs residues by artificial antibodies designed using a computational approach[J].Biomaterials,2009,30(18):3205-3211.
[18] KATRICE A L,LANE C S,RAYMOND D M.Molecular dynamics simulations of alkylsilane stationary-phase order and disorder[J].Anal Chem,2005,77(24):7862-7871.
[19] IMREA A R,KRASKA T.Estimation of spinodals from the density profile of the vapor—liquid interface[J].Fluid Phase Equilibria,2009,284(1):31-37.
Molecular dynamics simulation on adsorption behavior of 1-(2-hydroxyethyl)-2-alkyl-imidazoline corrosion inhibitor at Fe surface
ZHANG Jun1,JIANG Juan-juan2,REN Zhen-jia1,YU Wei-zhao3,YU Li-jun1,QIAO Gui-min1
(1.College of Physics Science and Technology in China University of Petroleum,Dongying257061,China;2.Offshore Oil Engineering(Qingdao)Corporation Ltd,Qingdao266520,China;3.College of Petroleum Engineering in China University of Petroleum,Qingdao266555,China)
The adsorption behaviors of six kinds of 1-(2-hydroxyethyl)-2-alkyl-imidazoline corrosion inhibitors with different chain length on Fe(001)surface were studied in liquid conditions by molecular dynamics simulation method.The results show that all polar head groups of six corrosion inhibitors attached to the Fe surface,and the imidazoline rings were nearly paralleled to the surface.The alkyl chains located in the liquid phase and inclined toward the Fe surface with a certain angle.With the increase of the chain length,the binding stabilization of inhibitor molecules on the Fe surface gradually strengthened and the compactness of inhibitor monolayer also enhanced.The inhibition performances of six inhibitor molecules by theoretical method agree well with experimental results.
adsorption;molecular dynamics;simulation;imidazoline corrosion inhibitor;Fe surface
TG 174.42;O 646
A
10.3969/j.issn.1673-5005.2010.05.029
1673-5005(2010)05-0159-05
2010-04-18
中国石油中青年创新基金项目(07E1021);山东省自然科学基金项目(Y2006B35)
张军(1968-),男(汉族),山东定陶人,副教授,博士,研究方向为腐蚀防护。
(编辑 刘为清)
