β-Diketones at Water/Supercritical CO2 Interface: A MolecularDynamics Simulation*
2009-05-15LIUShuyan刘淑延CHAIJingchun柴景春andYANGXiaoning杨晓宁
LIU Shuyan (刘淑延), CHAI Jingchun (柴景春) and YANG Xiaoning (杨晓宁),**
β-Diketones at Water/Supercritical CO2Interface: A MolecularDynamics Simulation*
LIU Shuyan (刘淑延)1, CHAI Jingchun (柴景春)2and YANG Xiaoning (杨晓宁)2,**
1School of Mechanical and Power Engineering, Nanjing University of Technology, Nanjing 210009, China2State Key Laboratory of Material-Orientated Chemical Engineering, Nanjing University of Technology, Nanjing 210009, China
The structural and dynamical properties of hexafluoroacetylacetone (HFA) and acetylacetone (AA) at the water/supercritical CO2(Sc-CO2) interface at 20 MPa and 318.15 K are investigated by molecular dynamics simulations. The TIP3P potential is used for water and the EPM2 model is for CO2. The water phase and SC-CO2phase form a distinct immiscible liquid-liquid interface. The two chelating molecules show interfacial preference. Comparatively, the AA molecules show somewhat more preference for interfacial region, whereas the HFA molecules are preferably near the Sc-CO2phase. The orientational distribution of the β-diketone molecules and the radial distribution functions between β-diketones and solvents are obtained in order to study the microscopic structural properties of the β-diketones at the water-SC-CO2interface. It is found that the translational diffusion and rotational diffusion of HFA and AA are obviously anisotropic and decrease as the β-diketone molecules approach the interface. The anisotropic dynamic behavior for the solute molecules is related to the corresponding structural properties.
molecular dynamics simulation, liquid/liquid interface, supercritical carbon dioxide, β-diketone
1 INTRODUCTION
Extraction and separation of metal ions from an aqueous phase using the supercritical CO2(SC-CO2) is emerging as a promising “green” technology. Compared with the conventional solvent extraction, supercritical fluid extraction is relatively fast and environmentally friendly. The solvency of Sc-CO2can be controlled effectively by adjusting temperature and pressure. Environmental applications, such as the extraction of heavy metals or radionuclides from contaminated soil or the metal removal from industrial waste water, have been the major focus in recent years [1-3].
For the low polarity of Sc-CO2solvent, the charge neutralization of metal ions in the Sc-CO2phase requires chelating agents [4]. A critical aspect is the design and synthesis of the chelating agents [5]. The β-diketone, as an important class of chelating acidic ligand, has been widely used in extraction processes [3, 4, 6-8]. These β-diketones are commercially available and inexpensive. Under appropriate conditions, the enolic hydrogen atom of the β-diketone ligand can be replaced by a metal ion to produce a chelate ring, thereby shifting the keto-enol equilibrium in favor of the enol form [6].
In the SC-CO2extraction of metals from aqueous phases, the chemical reactions occurring at the water/Sc-CO2interface may be fast compared to the mass transfer processes [9, 10]. Therefore, the interfacial properties for solutes to occur at the water/Sc-CO2interface are of great importance. However, the detailed microscopic knowledge of the interfacial properties of chelating agent at the water/Sc-CO2interface, both experimental and theoretical, is still limited. The interface between the two immiscible fluids possesses many unique properties, different from those in the two bulk phases. For common liquid-liquid systems, some microscopic properties can be measured using the modern experimental methods [11-16], however, the experimental probing of the microscopic interfacial properties for the water/Sc-CO2interface under high pressure condition remains extremely difficult.
Molecular dynamics (MD) simulation has become a powerful tool for the study of interfacial properties and it can provide complementary information on the microscopic structural and dynamical properties of liquid-liquid interface [17-22], serving as a source of complementary information and a guide to experiment. These properties are vital for process design and operation. For the water/Sc-CO2interface, Rocha and Johnston [23] have investigated the molecular structure of the neat water/Sc-CO2interface. Moreover, some complex systems have been studied, including ionic species, surfactants and ligands at the water/Sc-CO2interface [24-30]. However, to our knowledge, the detailed interfacial properties on molecule level about the chelating agents, hexafluoroacetylacetone (HFA) and acetylacetone (AA), at the water/Sc- CO2interface remains unresolved.
In this study, we use MD simulation to investigate the structural behavior and dynamic properties of the enol form of AA and its fluorinated analog, HFA, at the water/Sc-CO2interface. The simulation is at 318.15 K and 20 MPa.

Table 1 Structural parameters for β-diketones
2 MODEL AND METHOD
2.1 Potential models
In this work, we use the TIP3P potential for the water molecules [31]. For CO2, the EPM2 model of Harris and Yung [32] is adopted, which consists of 3 Lennard-Jones sites and three point charges located in the centers of the LJ sites. The β-diketones, CF3COCHCOHCF3and CH3COCHCOHCH3are fully optimized with the density functional theory (DFT) approach, where the generalized gradient approximation GGA functional (GGA-PW91 [33]) is applied. The molecular structures of HFA and AA and the charge of every atom or group are shown in Fig. 1. The structure parameters derived from the optimization are given in Table 1.

Figure 1 Schematic structures and charges for two β-diketone molecules
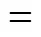
In this MD simulation, the total energy of the simulated system is the sum of the intramolecular and intermolecular interactions.
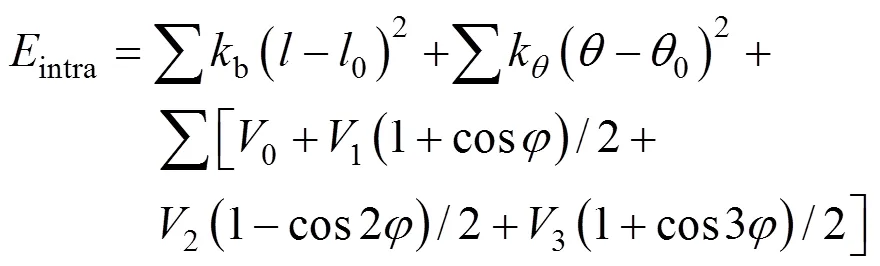
The intermolecular interactions between nonbonded pairs are given by
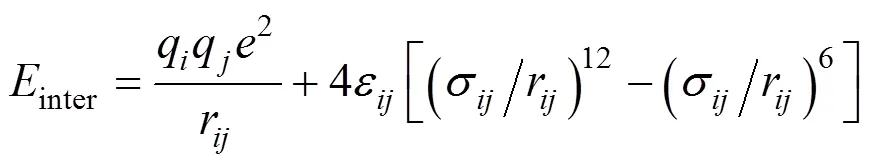
whereqandqare the charges centered on the individual particles of different molecules,ris the distance between the sitesand,σandεare the size and energy parameters of the LJ potential. The potential parameters of the HFA molecule are from Ref. [34], and those of the AA ligand are from the OPLS potential model [35]. The LJ parameters for the interactions between dissimilar particles are calculated using the Lorentz-Berthelot mixing rule.
In order to testify the rationality of the potential model of HFA, we calculate the diffusion coefficient of HFA in SC-CO2under various conditions (at 318.15 K and 10.62-22.20 MPa). The results are listed in Table 2. A relatively close agreement is observed between the simulation results and the experimental data [36], which may provide a measure of validity of the used potential model.
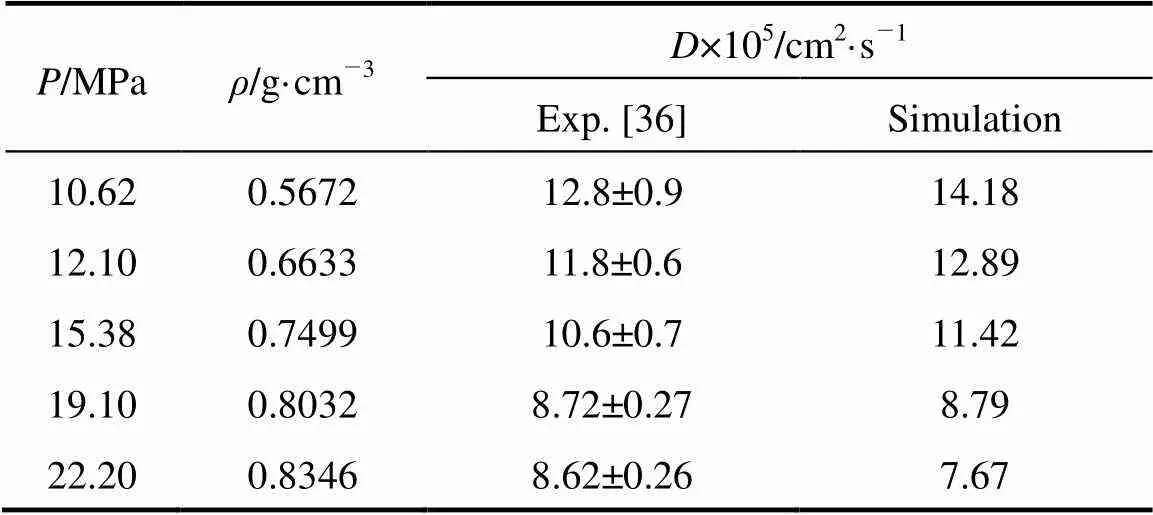
Table 2 Diffusion coefficient of HFA in supercritical CO2
2.2 Simulation method
First, two independent bulk solvent phases are set up. The initial bulk system for water phase contains 792 water molecules (LL2.8 nm,L13.02 nm) with1.0 g·cm-3, and the pure CO2bulk phase consists of 324 EPM2 CO2molecules (LL2.8 nm,L23.63 nm) with0.8314 g·cm-3, corresponding to the experimental density of the CO2phase at 318.15 K and 20 MPa. Then, the two bulk phases are connected to form the water/Sc-CO2interface with the cross section12L×L. The bulk water phase is in the negativedirection relative to the EPM2 CO2phase. The length of the initial simulation box indirection isLL1+L26.65 nm, and the initial total volume of the constructed system is×L.
In the MD simulation, the planar-enol form is adopted as the structure for the HFA and AA ligands based on the arguments in the literature [27] and the cited references herein. Although no experimental solubility data is available for the β-diketones, very low solubility (10-8-10-3in mole fraction) for their metal complexes [37, 38] may suggest a very limited solubility for the HFA and AA ligand molecules. Accordingly, only five chelating agent molecules are involved in the simulation systems.
Five β-diketone molecules are randomly distributed in the vicinity of the interface. In order to check the effect of initial position of β-diketone molecules on the finial equilibrium configuration, we also conduct a series of additional MD simulations by placing the β-diketone molecules at different initial positions, for example, in the bulk water phase and in the bulk CO2phase, respectively. These simulation tests show that after equilibrium, the ligand molecules are almost located at the same position in the interfacial region. Before the MD simulation runs, we carry out the energy optimization of the system. After the energy optimization, the first part of the equilibration simulation of 300 ps is performed at 318.15 K and 20 MPa in the NPT ensemble with a time step of 2 fs. The temperature is controlled with the Berendsen temperature bath, and a constant pressure is maintained using the Berendsen pressure bath. The Ewald summation method is used for the electrostatic interaction. In the simulation, the cell volume is changed along the direction ofL, whileLandLremain constant. The volume fluctuation ensures that the pressure is stabilized and oscillates around their required value (20 MPa). Periodic boundary conditions are applied in all directions. The final lengths of simulation boxes indirection for all systems investigated are in the range of 6.56-6.75 nm. After the first equilibrium NPT MD run, a second equilibration run for 200 ps a performed in an NVT ensemble. The production MD run is carried out for the next 1000 ps in the NVT ensemble. A total of 1.5 ns is for each MD simulation.
3 RESULTS AND DISCUSSION
3.1 Density profile
The density profile along the interface normal (-direction) is shown in Fig. 2. The profiles are calculated using the slabs of thickness 0.06 nm parallel to the interfacial plane, and thecoordinates are defined with respect to the center of mass in the systems. Fig. 2 indicates that the potential models of water and CO2are appropriate for describing the immiscible liquid- liquid interface formation of the water/Sc-CO2system, which is similar with the simulation study of the net water/Sc-CO2interface [23]. There exists a limit in the mutual solubility of water and CO2. Only limited CO2molecules are dissolved in water, whereas the CO2phase usually does not contain water molecules.
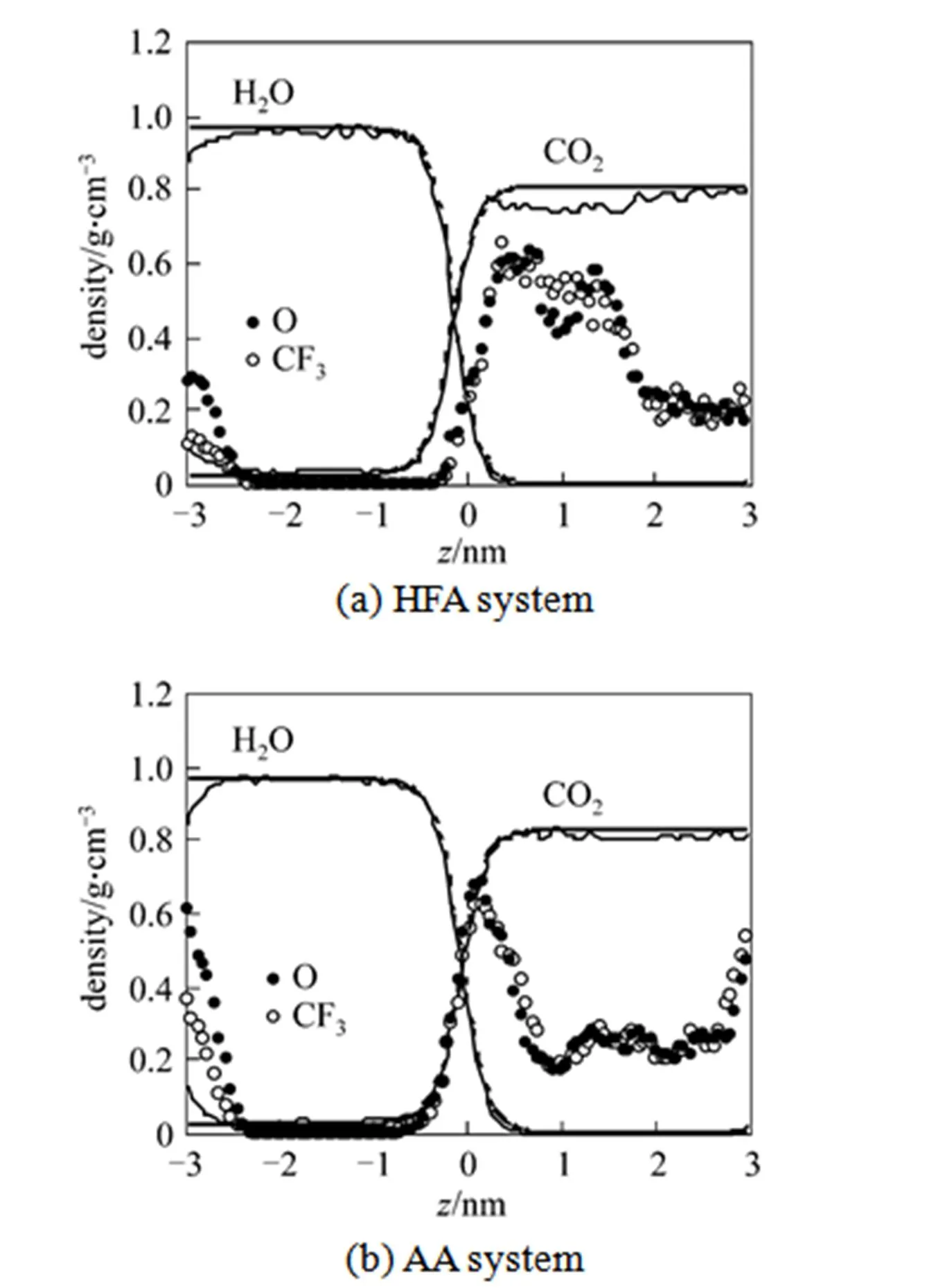
Figure 2 Density profiles of two β-diketone ligands at the water/Sc-CO2interface along the interface normal axis
(Dashed lines: fitted density profiles; O: oxygen atom of HFA and AA molecules; CF3/CH3: CF3/CH3groups in HFA and AA molecules)
Figure 2 shows that, after equilibrium, both HFA and AA molecules are concentrated in the region near the water/Sc-CO2interface. Comparatively, the AA molecules show somewhat more preference for the interfacial region, whereas the HFA molecules are preferably near the SC-CO2phase due to its CO2-philic feature. This is consistent with the fact that the F-substituted β-diketone molecule prefers the CO2environment. Our simulation result is qualitatively consistent with the previous simulation of X-aryls in the water/Sc-CO2interface [39]. In this previous study, the fluorinated benzene molecules also stay away from the water/Sc-CO2interface and they do not have a preference for interfacial position, whereas benzene usually remains close to the water/Sc-CO2interface [39]. In our simulation, almost no β-diketone molecules are found in the aqueous phase due to their insolubility in the aqueous phase. A similar qualitative observation has also been found in the MD simulation study of Galand and Wipff [26].
3.2 Structure properties
3.2.1
The distribution of cosis shown in Fig. 3, where the orientation curve with a little noise is obtained due to the smaller number of molecules used to make the simulation average. For the HFA molecule, the distribution of cosin region A indicates that the vector from the C1atom to O4atom of the β-diketone molecules is aligned preferentially perpendicular to the interface, that is, the solute molecules might adopt the favorable orientations with the hydrophilic group toward the aqueous phase. However, in region B, the distribution is relatively uniform, suggesting no preferential orientation. For the AA molecule, a similar orientation behavior is observed. The occurrence of the distribution maximum point implies that the AA molecule orientation possesses a specified orientation.

Figure 3 Probability distribution of cos
◆ interfacial layer (region A);○ region B
The distribution ofis not shown here. For HFA and AA, the orientation of 60º≤≤90º is preferred near the interface (region A), indicating that the plane of the β-diketone molecule is inclined to be somewhat perpendicular to the interface. The distribution ofis also not uniform in region B due to the weak effect of the interface.
3.2.2
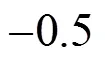
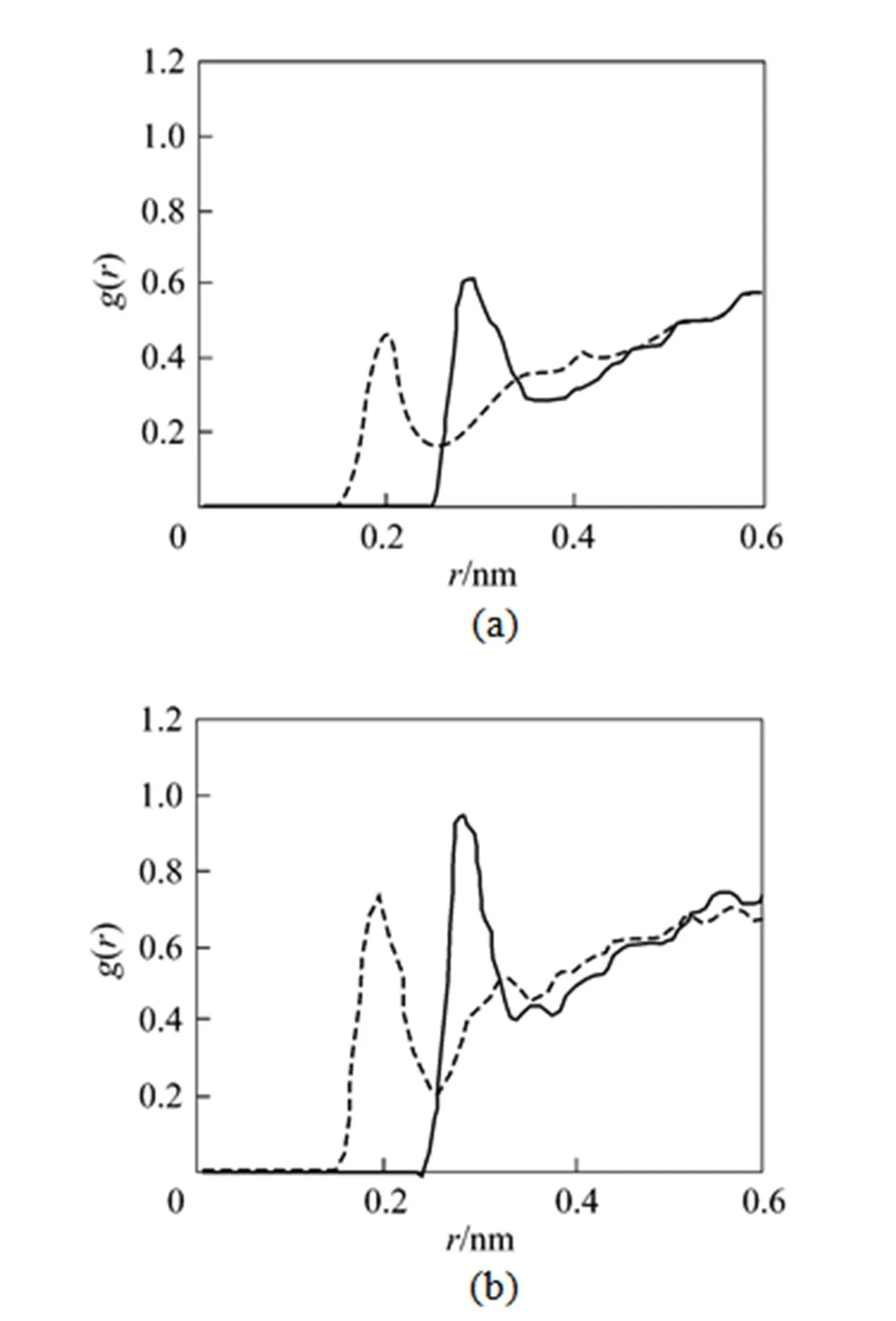
Figure 4 (a) RDFs between HFA and water; (b) RDFs between AA and water

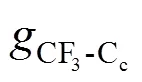
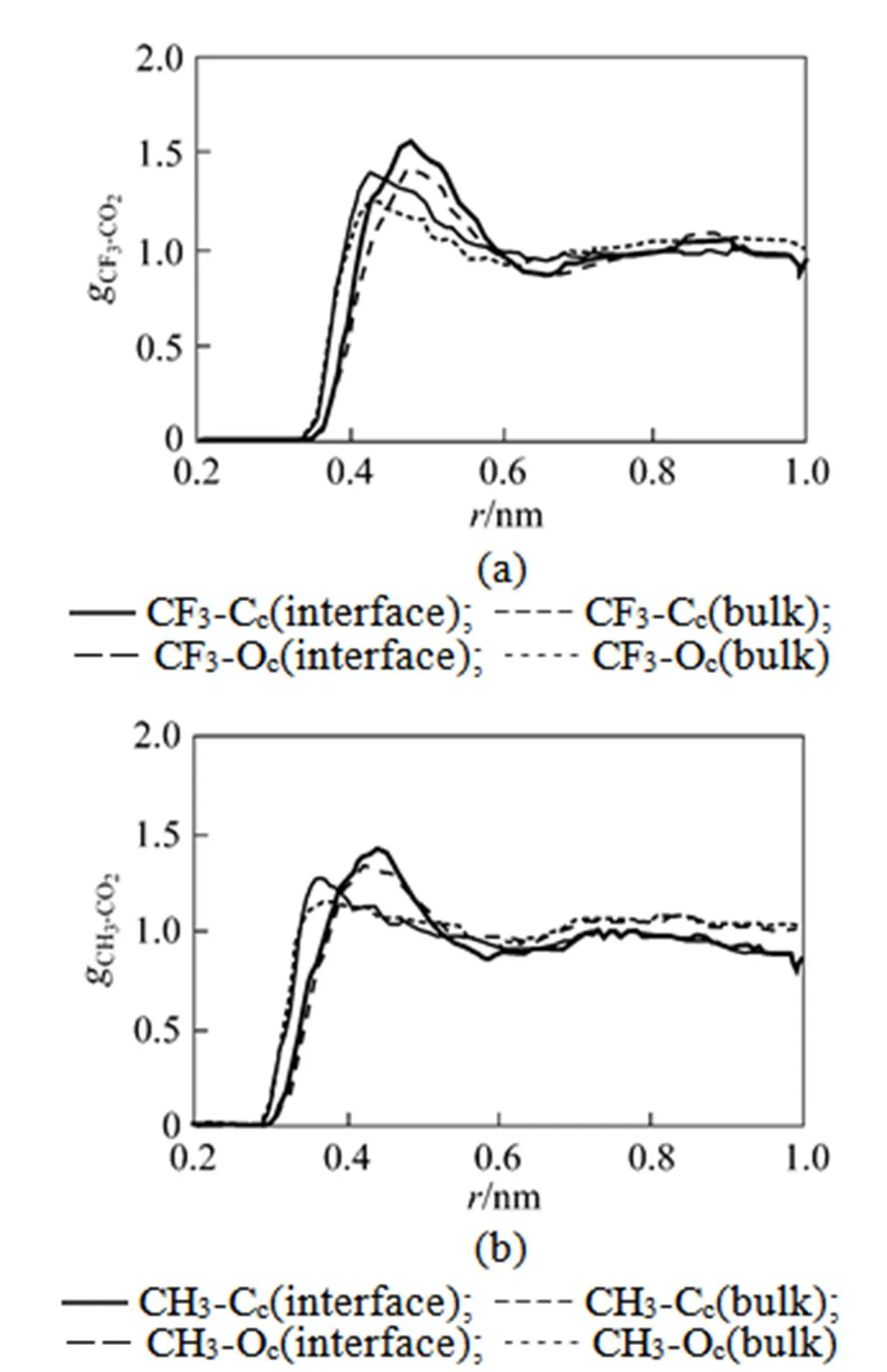
Figure 5 (a) RDFs between HFA and CO2; (b) RDFs between AA and CO2
The total solvation coordination number around the β-diketone molecules is estimated based on the RDF between the mass center of the β-diketone and CO2molecules. For the HFA and AA molecules, the solvation radius is chosen to be the first minimum position beyond the first peak in the corresponding RDFs. Thus, all the solvent molecules, including CO2and water, are considered to be located in the first solvation shell of solute molecule when the distance between the solvent and solute molecules is lower than the corresponding solvation radius. Fig. 6 shows the coordination number of HFA and AA as a function of the distance relative to the interface. The coordination number is enhanced significantly from the bulk CO2phase to the interfacial region. At the interfacial region, the presence of water molecules, partially replacing the CO2molecules, enlarges the solvating layer structures around the solute.
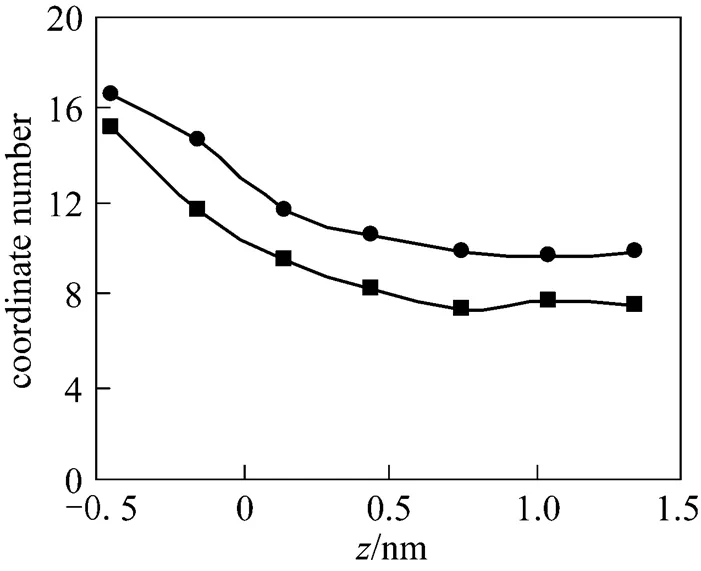
Figure 6 The total solvent coordination number of HFA and AA as a function ofcoordinate relative to the interface
● AA;■ HFA

Figure 7 MSDs of β-diketones as a function of the distance from the interface for HFA and AA

3.3 Dynamic behavior
3.3.1
In order to evaluate the diffusion behavior of the chelating agents in the interfacial region, the three components of the mean square displacements (MSD) for the chelating agents are calculated. The MSDs of/anddirections of the HFA and AA molecules, at various positions from the interface, as a function of time are shown in Fig. 7. The MSDs are calculated using the slab of 0.5 nm thickness in relative to the interface. We monitor thecoordinate of every molecule in order to prevent the statistical accumulation of data from molecules diffusing outside the slab during the interval [,+], whereis the time origin andis the time increment, which is not greater than half the average residence time of the molecules in each slab. This calculation procedure has been successfully applied to the previous study on liquid-liquid interface [19]. In our calculation, the mean residence timeis estimated to be 10 ps. Fig. 7 shows that the slope of the MSD lines decreases near the interfacial region, suggesting a reduced diffusion phenomenon near the interface. This behavior is related to the total solvation structure enhancement from the CO2bulk region to the interfacial region (see Fig. 6). The enhancement may lead to a stronger blocking effect on the solute movement at the interfacial region. An obvious anisotropic behavior is observed for the diffusion of HFA and AA. The diffusion coefficient perpendicular to the interface is smaller than that in the direction parallel to the interface. This is caused by the anisotropic environment that hinders the solute movement toward the interface.
Figure 8 Diffusion coefficients of CO2and water as a function of the distance from the interface
■Dof H2O;□Dof H2O;◆Dof CO2;◇Dof CO2
The diffusion anisotropy of solvent near liquid- liquid interface has been studied extensively through molecular simulations [19, 43, 44]. In order to further examine the diffusion behavior near the water/Sc-CO2interface, we also investigate the diffusion for the two solvents based on the Einstein approach [19]. Fig. 8 gives the parallel and perpendicular diffusion coefficient profiles of CO2and water from the bulk phase to the interface region. For the two solvents, the self-diffusion coefficients also show an anisotropic trend, that is, the diffusion coefficient perpendicular to the interface is smaller than the parallel one. No obvious difference is observed in the diffusion anisotropy of solvent with the presence of chelating agents. For CO2, there is a reduction of the anisotropic diffusion near the interface, while for the water an enhancement of the anisotropy diffusion is observed at the interface. This is expected because near the interface the hydrogen network structure of water molecules becomes weaker [23] and a possible specific attractive interaction between CO2and water molecules may restrict the CO2molecule movement.
3.3.2
The orientational relaxation time and rotational diffusion coefficients of the two chelating agents in the water/Sc-CO2interface are investigated. Based on the Debye theory of angular relaxation, the orientation correlation equations are as follows [45],
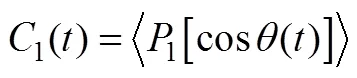
Pl is an l-order Legendre polynomial [45]. In this study, is chosen for the orientation dynamics. , in which li is a particular unit vector. In this study, the unit vector from the C1 atom to the O4 atom is adopted as l1 in the orientation calculation. The rotational diffusion coefficient DR1 is calculated by
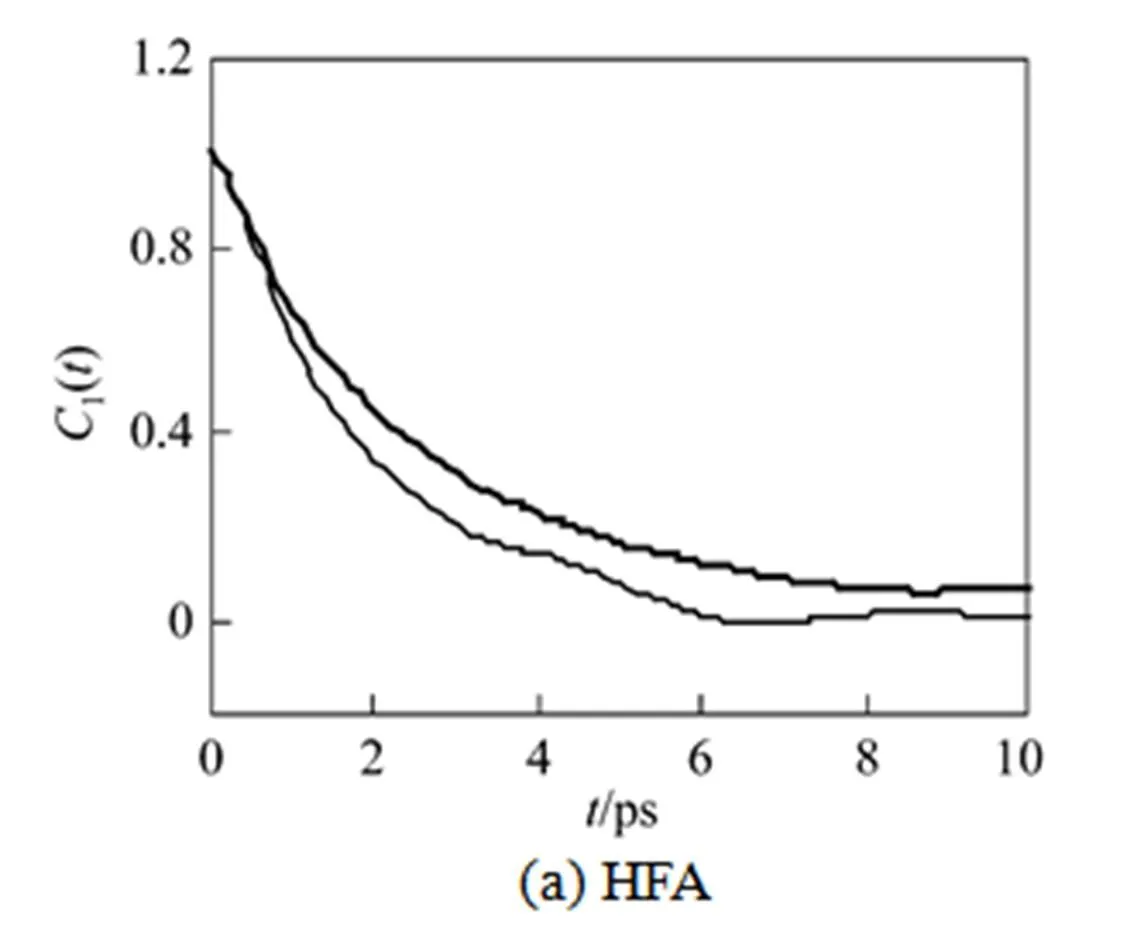
Figure 9 Orientational correlation functions of β–diketones at the interface and in the CO2bulk phase


Rotational diffusion coefficients are also shown in Table 3. The rotational diffusion coefficients of the β-diketones at the water/Sc-CO2 interface system are in the range of 0.1-0.3 ps-1, which are noticeably greater than the rotational diffusion coefficient of surfactin molecule at the hexane/water interface (0.02-0.03 ps-1) [48]. This is due to the fact that the β-diketone molecule is much smaller in size than the surfactin molecule. The rotational diffusion coefficients of the β-diketones at the region near the interface are smaller than those in bulk CO2 phase. This reduced rotation diffusion has been observed for surfactin molecules at the hexane/water interface [48]. The slowdown in the rotational diffusion at the interface can be also ascribed to the enhancing solvation structure around the solute molecules, which imposes a constraint for the rotational movement at the interfacial region.
4 CONCLUSIONS
We have studied the structural and dynamical properties of two β-diketone chelating agents, hexafluoroacetylacetone (HFA) and acetylacetone (AA), at the water/Sc-CO2interface system by the MD simulations.
The density profiles of the two β-diketone molecules are obtained. The two solute molecules are concentrated in the vicinity of the water/Sc-CO2interface. The simulation result further indicates that the AA molecule shows relatively stronger interfacial preference, whereas the HFA molecules are preferably near the bulk SC-CO2phase due to its CO2-philic feature. After introducing the β-diketone molecules to the water/Sc-CO2system, the immiscible water/Sc-CO2interfacial structure does not change significantly. In the interfacial region, the HFA and AA molecules show preferential orientations to some extent due to the favorable interaction between the hydrophilic group and the aqueous phase. The radial distribution functions between the β-diketone molecules and the solvents are examined and it is found that the strength of AA molecules forming hydrogen bond with aqueous phase is greater than that of HFA. Their structural behaviors are consistent with the obtained density distributions in the interfacial region.
The diffusion behavior of HFA and AA is found to be anisotropic. The diffusion coefficients perpendicular to the interface are smaller than those in the direction parallel to the interface. The reduced mobility of the solute molecules at the interface is related to the change of their solvation structure from the bulk phase to the interface phase. There is also an anisotropic phenomenon for the solvent diffusion in the water/ Sc-CO2system. Near the interface, the enhancing solvation structure around the solute molecules results in a decrease in the rotational diffusion coefficient of the β-diketone molecules, which is consistent with the increasing orientational relaxation time at the interface.
NOMENCLATURE
l() orientational correlation function
R1rotational diffusion coefficients
unit charge
b,,0bond stretching force constant, actual bond length, equilibrium bond length
k,,0bond angle bending force constant, actual bond angle, equilibrium bond angle
a particular unit vector
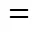
q,qcharges of sitesand
rdistance between the sitesand
0,1,2,3torsional force constants
εenergy parameter
σsize parameter
1orientational correlation time
torsional angle [Eq. (1)]
1 Clifford, Y.T., You, G.S., “Fractional of metal ions from water using supercritical carbon dioxide”,., 50 (7), 1627-1630 (2004).
2 Iwao, S., El-Fatah, S.A., Furukawa, K., Seki, T., Sasaki, M., Goto, M., “Recovery of palladium from spent catalyst with supercritical CO2and chelating agent”,.., 42, 200-204 (2007).
3 Chou, W.L., Yang, K.C., “Effect of various chelating agents on supercritical carbon dioxide extraction of indium (III) ions from acidic aqueous solution”,..., 154, 498-505 (2008).
4 Chen, J.L., Liu, C.Y., “Chelating resin sorption followed by supercritical fluid extraction and liquid chromatographic determination of aluminum in liquid samples”,.., 494, 125-132 (2003).
5 Shamsipur, M., Ghiasvand, A.R., Yamini, Y., “Extraction of uranium from solid matrices using modified supercritical fluid CO2”,.., 20, 163-169 (2001).
6 Ozel, M.Z., Burford, M.D., Clifford, A.A., Bartle, K.D., Shadrin, A., Smart, N.G., Tinker, N.D., “Supercritical fluid extraction of cobalt with fluorinated and non-fluorinated β-diketones”,.., 346, 73-80 (1997).
7 Lalntz, K.E., Tachlkawa, E., “Extraction of lanthanides from acidic solution using tributyl phosphate modified supercritical carbon dioxide”,.., 66, 2190-2193 (1994).
8 Murphy, J.M., Erkey, C., “Thermodynamics of extraction of copper (II) from aqueous solutions by chelation in supercritical carbon dioxide”,..., 31, 1674-1679 (1997).
9 Erkey, C., “Supercritical carbon dioxide extraction of metals from aqueous solutions: A review”,.., 17, 259-287 (2000).
10 Murphy, J.M., Erkey, C., “Copper (II) removal from aqueous solutions by chelation in supercritical carbon dioxide using fluorinated beta-diketones”,...., 36, 5371-5376 (1997).
11 Mitrinovic, D.M., Zhang, Z., Williams, S.M., Huang, Z., Schlossman, M.L., “X-ray reflectivity study of the water-hexane interface”,..., 103, 1779-1782 (1999).
12 Zarbakhsh, A., Querol, A., Bowers, J., Yaseen, M., Lu, J.R., Webster, J.R.P., “Neutron reflection from the liquid-liquid interface: Adsorption of hexadecylphosphorylcholine to the hexadecane-aqueous solution interface”,, 21, 11704-11709 (2005).
13 Ohe, C., Ida, Y., Matsumoto, S., Sasaki, T., Goto, Y., Noi, M., Tsurumaru, T., Itoh, K., “Investigations of polymyxin B-phospholipid interactions by vibrational sum frequency generation spectroscopy”,..., 108, 18081-18087 (2004).
14 Nochi, K., Yamaguchi, A., Hayashita, T., Uchida, T., Teramae, N., “Direct observation of alkali metal ion recognition processes at the heptane/water interface by second harmonic generation spectroscopy”,..., 106, 9906-9911 (2002).
15 Takechi, H., Adachi, K., Monjushiro, H., Watarai, H., “Linear dichroism of Zn(II)-tetrapyridylporphine aggregates formed at the toluene/water interface”,, 24, 4722-4728 (2008).
16 Timgren, A., Tragardh, G., Tragardh, C., “Application of the PIV technique to measurements around and inside a forming drop in a liquid-liquid system”,., 44, 565-575 (2008).
17 Chang, T.M., Dang, L.X., “Molecular dynamics simulations of CCl4-H2O liquid-liquid interface with polarizable potential models”,..., 104, 6772-6783 (1996).
18 Benjamin, I., “Theoretical study of the water/l,2-dichloroethane interface: Structure, dynamics, and conformational equilibria at the liquid-liquid interface”,..., 97, 1432-1445 (1992).
19 Fernandes, P.A., Cordeiro, M.N.D.S., Gomes, J.A.N.F., “Molecular dynamics simulation of the water/2-heptanone liquid-liquid interface”,..., 103, 6290-6299 (1999).
20 Jedlovszky, P., Vincze, A., Horvai, G., “Full description of the orientational statistics of molecules near to interfaces. Water at the interface with CCl4”,...., 6, 1874-1879 (2004).
21 Jorge, M., Natalia, M., Cordeiro, D.S., “Intrinsic structure and dynamics of the water/nitrobenzene interface”,..., 111, 17612-17626 (2007).
22 Buhn, J.B., Bopp, P.A., Hampe, M.J., “Structural and dynamical properties of liquid-liquid interfaces: A systematic molecular dynamics study”,..., 125, 187-196 (2006).
23 da Rocha, S.R.P., Johnston, K.P., Westacott, R.E., Rossky, P.J., “Molecular structure of the water/supercritical CO2interface”,..., 105, 12092-12104 (2001).
24 Vayssiere, P., Wipff, G., “Ethers, crown ethers and 18-crown-6 K + complexes at a water/SC-CO2interface: A molecular dynamics study”,...., 5, 127-135 (2003).
25 Schurhammer, R., Berny, F., Wipff, G., “Importance of interfacial phenomena in assisted ion extraction by supercritical a molecular dynamics investigation”,...., 3, 647-656 (2001).
26 Galand, N., Wipff, G., “Uranyl extraction by β-diketonate ligands to SC-CO2: Theoretical studies on the effect of ligand fluorination and on the synergistic effect of TBP”,..., 109, 277-287 (2005).
27 da Rocha, S.R.P., Johnston, K.P., Rossky, P.J., “Surfactant-modified CO2-water interface: A molecular view”,..., 106, 13250-13261 (2002).
28 Chaumont, A., Galand, N., Schurhammer, R., Vayssiere, P., Wipff, G., “Accumulation of host-guest ion complexes with different counterions at the water/supercritical CO2interface: A molecular dynamics study”,..., 53, 1459-1565 (2004).
29 Guo, J.X., Sun, S.X., Yuan, S.L., Ran, X.K., Xu, G.Y., “Computer simulations of extractant primary amine N1923 and N1923 hydrochloride salt at water/chloroform interface”,.., 32, 451-455 (2006).
30 Lu, L.Y., Berkowitz, M.L., “The effect of the rigidity of perfluoropolyether surfactant on its behavior at the water/supercritical carbon dioxide interface”,..., 109, 21725-21731 (2005).
31 Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W., Klein, M.L., “Comparison of simple potential functions for simulating liquid water”,..., 79, 926-935 (1983).
32 Harris, J.G., Yung, K.H., “Carbon dioxide's liquid-vapor coexistence curve and critical properties as predicted by a simple molecular model”,..., 99, 12021-12024 (1995).
33 Perdew, J.P., Chevary, J.A., Vosko, S.H., Jackson, K.A., Pederson, M.R., Singh, D.J., Fiolhais, C., “Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation”,.., 46, 6671-6687 (1992).
34 Collazo, N., Shin, S., Rice, S.A., “Molecular-dynamics studies of the structure and properties of monolayers of perfluorinated amphiphiles”,..., 96, 4735-4742 (1992).
35 Jorgensen, W.L., Briggs, J.M., Contreras, M.L., “Relative partition coefficients for organic solutes from fluid simulations”,..., 94, 1683-1686 (1990).
36 Yang, X.N., Matthews, M.A., “Diffusion of chelating agents in supercritical CO2and a predictive approach for diffusion coefficients”,..., 46, 588-595 (2001).
37 Lagalante, A.F., Hansen, B.N., Bruno, T.J., Sievers, R.E., “Solubilities of copper (II) and chromium (III) beta-diketonates in supercritical carbon-dioxide”,.., 34, 5781-5785 (1995).
38 Ozel, M.Z., Bartle, K.D., Clifford, A.A., Burford, M.D., “Extraction, solubility and stability of metal complexes using stainless steel supercritical fluid extraction system”,.., 417, 177-184 (2000).
39 Galand, N., Wipff, G., “Solvation of benzene derivatives in SC-CO2: A molecular dynamics study of fluorination effects”,.., 27, 1319-1325 (2003).
40 Guardia, E., Marti, J., Padro, J.A., Saiz, L., Komolkin, A.V., “Dynamics in hydrogen bonded liquids: Water and alcohols”,..., 96-97, 3-17 (2002).
41 Higashi, H., Iwai, Y., Miyazaki, K., Arai, Y., “Molecular dynamics simulation of fluorination effect for solvation of trifluoromethylbenzoic acid isomers in supercritical carbon dioxide”,.., 31, 725-730 (2005).
42 Meredith, J.C., Johnston, K.P., Seminario, J.M., Kazarian, S.G., Eckert, C.A., “Quantitative equilibrium constants between CO2and lewis bases from FTIR spectroscopy”,..., 100, 10837-10848 (1996).
43 Wang, H., Carlson, E., Henderson, D., Rowley, R.L., “Molecular dynamics simulation of the liquid-liquid interface for immiscible and partially miscible mixtures”,.., 29, 777-785 (2003).
44 Michael, D., Benjamin, I., “Solute orientational dynamics and surface roughness of water/hydrocarbon interfaces”,..., 99, 1530-1536 (1995).
45 Inomata, H., Saito, S., Debenedetti, P.G., “Molecular dynamics simulation of infinitely dilute solutions of benzene in supercritical CO2”,., 116, 282-288 (1996).
46 Smirnov, K.S., Bougeard, D., “A molecular dynamics study of structure and short-time dynamics of water in kaolinite”,..., 103, 5266-5273 (1999).
47 Siavosh-Haghighi, A., Adams, J.E., “Rotational relaxation in a nondipolar supercritical fluid: Toluene in CO2”,..., 105, 2680-2686 (2001).
48 Nicolas, J.P., “Molecular dynamics simulation of surfactin molecules at the water-hexane interface”,.., 85, 1377-1391 (2003).
2008-10-15,
2009-07-07.
the National Natural Science Foundation of China (20776066, 20476044) and the Specialized Research Fund for the Doctoral Program of Higher Education of China (20060291002).
** To whom correspondence should be addressed. E-mail: yangxia@njut.edu.cn
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Study on the Flow Field around Two Parallel Moving Bubbles andInteraction Between Bubbles Rising in CMC Solutions by PIV*
- Kinetic Rate Constant of Liquid Drainage from Colloidal Gas Aphrons*
- Analysis of Sucrose Esters with Long Acyl Chain by Coupling ofHPLC-ELSD with ESI-MS System*
- Adsorptive Removal of Copper Ions from Aqueous Solution UsingCross-linked Magnetic Chitosan Beads
- Gas Flow in Unilateral Opening Pulse Tubes Based on Real Gas Equation of State
- Solubility of Piperine in Supercritical and Near Critical Carbon Dioxide*