4′-N,N-二乙氨基-3-羟基苯并黄酮激发态分子内质子转移机制的溶剂极性效应
2025-02-20姜羊林陈明卿梁民尧奕歌张燕王鹏张建平
关键词:4′-N,N-二乙氨基-3-羟基苯并黄酮;激发态分子内质子转移;密度泛函理论/含时密度泛函理论;溶剂极性效应;荧光动力学
1 引言
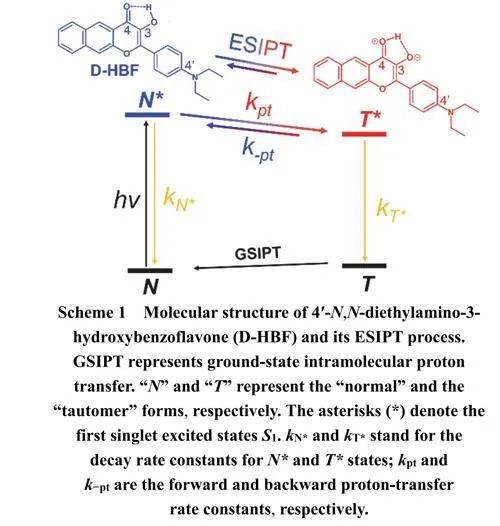
激发态分子内质子转移(ESIPT)是一种在化学和生物体系中重要的基础光化学反应,它主要发生在具有分子内氢键的发色团中。ESIPT可以用如示意Scheme 1所示的四能级模型描述:当分子被光激发后,通过Frank-Condon跃迁迅速生成分子构型不变的“Normal”激发态(N*态),N*态随后会通过激发态分子内质子转移(ESIPT)过程转化为N*态的“Tautomer”激发态(T*态)。通常情况下,N*和T*态可以通过双荧光发射的方式回到各自基态,而处于Tautomer基态(T态)的分子又可以通过基态分子内质子转移(GSIPT)反应回到Normal基态(N态) 1,最终完成四能级循环。
在具有ESIPT特性的分子中,3-羟基黄酮衍生物(3-HFs)因其来自天然,且具有对周围环境(主要是各种溶剂)变化敏感的荧光性质而引起人们广泛关注2,3。对3-HFs荧光特性的溶剂效应研究已被应用于各种领域,如:激光材料4、监测生物柴油中甲醇含量的比率型荧光探针5、监测脂滴形貌的动态荧光成像探针6等。
自20世纪90年代以来,对3-HFs的ESIPT机制的溶剂效应研究已广泛开展7–9。在非质子乙腈/苯混合溶剂中,研究者开展了对4′-N,N-二烷基氨基-3-羟基黄酮的ESIPT动力学的实验和理论研究10。Chou等人报道了从正庚烷转变为甲醇时,4′-N,N-二乙氨基-3-HF的N*态发射峰强度显著增加且大幅红移(gt; 3000 cm−1) 11,他们提出:在强极性、质子型溶剂乙醇中,4′-二乙氨基向羰基氧原子发生了分子内电荷转移,从而导致N*态具有两性离子构型。Shynkar等人研究揭示了4′-N,N-二乙氨基-3-HF的ESIPT反应(在两个激发态之间的快速可逆反应)在几十皮秒的尺度上发生12。Roshal等人探索了溶剂效应对4′-二甲基氨基-3-HF中ESIPT速率的影响,该分子可以在超快时间尺度上形成分子内电荷转移(ICT)态13。Douhal等人则在同一个化合物的研究中观察到其ICT与质子转移的竞争关系14:非极性溶剂中,ICT不会发生,只在2 ps时间内发生质子转移过程;极性溶剂中,ICT发生在100–200fs内,而质子转移则在几皮秒到几十皮秒内发生。此外,Chou等人使用飞秒荧光上转换技术并结合从头算,提出:不同溶剂导致4′-N,N-二乙氨基-3-HF的电子态势垒不同,从而使其N*和T*态之间存在很大平衡极化差异,最终影响正向和反向ESIPT速率15。Rumble等人使用飞秒时间分辨克尔门发射光谱技术研究4′-N,N-二乙氨基-3-HF在碳酸丙烯酯/乙腈混合溶剂中的ESIPT动力学,也得到类似结论16。Ghosh等人研究发现,在氢键溶剂中,由于4′-N,N-二甲基氨基-3-HF与溶剂会形成分子间氢键,从而导致激发态分子较慢的溶剂弛豫过程,测得~100 ps的ESIPT时间常数17。另一项研究表明,该化合物在三种不同烷基链长的正-烷基醇中也存在非常慢的ESIPT过程,并且ESIPT和N*溶剂化时间常数均随着烷基链长单调增加18。Kuang等人使用‘pump-dump-probe’光谱技术,揭示了4′-N,N-二乙氨基-3-HF存在超快的基态分子内质子转移(GSIPT)动力学过程,同时证明该过程是一个溶剂依赖的不可逆过程1。Chen等人使用DFT和TDDFT方法对4′-甲氧基-3-HF 3和4′-二甲基氨基-6,7-二甲氧基-3-HF 19的ESIPT溶剂效应进行了理论研究,发现随着溶剂极性增加,ESIPT逐渐变得困难,这与早期实验结果相一致。
已有研究表明苯并-3羟基黄酮(苯并-3HF)衍生物是一类在生物体系中具有潜在应用价值的一氧化碳释放分子(CORM) 20,21。4′-N,N-二乙氨基-3-羟基黄酮(D-HBF)则被最近的研究确认是一种高效的脂滴成像荧光探针6,因其具有与3-HF相似的ESIPT溶剂效应。此外,由于该衍生物在结构上具有扩展的π电子云体系,从而使其光谱响应范围变大。然而,关于D-HBF详细的ESIPT溶剂调节机制研究尚未见诸报道。
本研究采用多种光谱技术并结合理论计算,系统探讨了溶剂极性对D-HBF的ESIPT的调控机制。首先选择了三种极性依次增大、极化率基本相似的非质子型有机溶剂,分别为:环己烷(CYH)、乙醚(DEE)和四氢呋喃(THF)。实验和理论研究均表明溶剂极性会影响D-HBF的ESIPT反应平衡。4′-N,N-二乙氨基的引入导致N*态具有ICT特性,从而增强了其ESIPT动力学对溶剂极性的敏感性。溶剂极性增加会导致D-HBF正向和反向ESIPT反应能垒变大,从而导致相应的质子转移速率降低。此外,溶剂极性增加会降低N* T*吉布斯自由能变化的绝对值,最终导致平衡向N*态移动。本研究从实验和理论上系统地研究了溶剂极性对苯并黄酮衍生物的ESIPT机制影响,为其作为荧光探针的开发奠定了理论基础。
2 实验与理论计算
2.1 试剂
4-二乙氨基苯甲醛(98%)购于北京伊诺凯科技有限公司(中国北京),1-(3-羟基萘-2-基)乙酮(95%)来自药明康德(中国上海)。其他所有试剂和溶剂(gt; 99%)均来自北京通广精细化工公司(中国北京)。所有试剂均未经纯化,直接使用。
D-HBF 的合成和结构表征参考SupportingInformation 6。
2.2 实验方法
1H NMR使用Bruker AM 600 MHz谱仪(德国,卡尔斯鲁厄,布鲁克公司)测试。紫外-可见吸收光谱采用Carry 60 UV-Vis (美国,加利福尼亚州,安捷伦公司)分光光度计测试,荧光光谱使用FS5荧光光谱仪测量(英国,爱丁堡,爱丁堡仪器公司)。荧光发射光谱的激发波长(λex)为430 nm,测量时将λex处吸光度调至lt; 0.1。绝对荧光量子产率(ΦFL)使用FLS 980荧光光谱仪(英国,爱丁堡,爱丁堡仪器公司)测得(积分球)。D-HBF在三种溶剂中的荧光寿命(τ,0–50 ns范围)使用FLS980-STM荧光光谱仪(英国,爱丁堡,爱丁堡仪器公司)的时间相关单光子计数(TCSPC)模块结合爱丁堡仪器公司的EPL-405 ps脉冲二极管激光器作为激发光源测得。其中,待测样品的在420 nm处吸光度为0.02。仪器响应函数(IRF)用30%的硅胶-水悬浊液作为散射体,采用与样品相同的测试方法测得(120ps)。
D-HBF在0–600 ps时间尺度的荧光寿命(τ)使用条纹相机测定。通过钛蓝宝石放大器(Mira-900Coherent,美国)获得工作频率为76 MHz且脉冲持续时间约为100 fs的830 nm脉冲激光。该激光被分成两束光:一束用于触发条纹相机;另一束先经过BBO晶体倍频到415 nm,再通过脉冲选择器将其降低到3.8 MHz,用来激发样品。样品发出的荧光由条纹相机(C5680,滨松,日本)收集。其中,条纹相机IRF ~10 ps。
2.3 计算部分
所有理论计算均使用Gaussian 16程序22,采用密度泛函理论(DFT)和时间依赖的密度泛函理论(TD-DFT)方法,结合B3LYP-D3/6-311+g(d,p)基组进行计算。为更准确地计算弱静电力相互作用,使用DFT-D3进行色散校正。此外,采用自洽反应场(SCRF)中的极化连续介质模型(PCM)对相应的溶剂环境进行模拟。在构象优化过程中,分子的键长、键角或二面角没有施加任何限制。分别计算了D-HBF的N、N*、T和T*态红外振动频率和偶极矩,计算结果没有虚频存在,表明优化的结构代表能量最小值。为了研究ESIPT反应与氢键之间的关系,计算了D-HBF在S0和S1态下势能曲线,O―H键长以0.05 Å (1 Å = 0.1 nm)每步的增量变化了26步。
吉布斯自由能的值是根据数学公式G = E +ΔGcorr + ZPE进行矫正,其中ΔGcorr吉布斯自由能校正项,ZPE是零点能项22。
3 结果与讨论
3.1 UV-Vis吸收光谱和荧光光谱
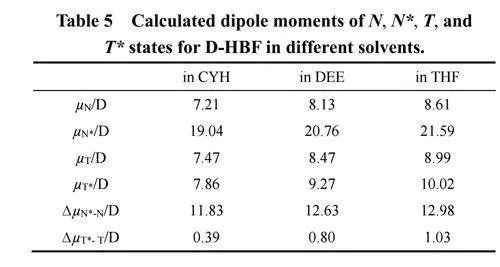
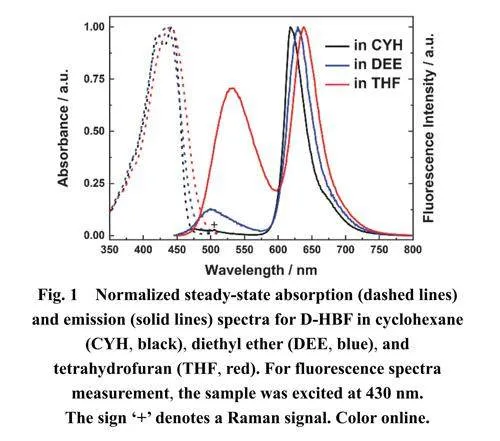
图1给出了室温下D-HBF在三种溶剂中的稳态吸收和发射光谱(结果归一化)。相应的光谱信息在总结于表1中。三种溶剂分别为:环己烷(CYH)、乙醚(DEE)和四氢呋喃(THF);它们具有相似的极化率,但极性有显著差异,Table S1中分别用溶剂的极化因子R(η)和极性因子P(ε)表示它们的差异,该表中另一个溶剂极性参数Δf 23,是基于溶剂的介电常数(ε)和折射率(η)计算得出。
三种溶剂中,D-HBF在350–470 nm范围内都呈现典型的π–π*跃迁吸收峰。其中,在CYH中的吸收光谱表现出清晰的振动结构,而在DEE和THF中,由于溶剂的极性效应导致精细结构消失。随着溶剂极性的增加,吸收峰逐渐展宽和红移。在所有溶剂中都观察到D-HBF的双荧光发射峰。450–600nm和600–750 nm范围内的发射带可分别归属为N*和T*发射(如示意图1所示)。随着溶剂极性的增加,N*和T*发射带均发生红移,其中N*发射比T*发射红移更为显著。有趣的是,溶剂极性的增加导致N*的斯托克斯位移相比于T*显著增加(见表1),表明N*能级对溶剂极性变化更加敏感。T*在三种溶剂中斯托克斯位移(约7000 cm−1)都异常大,结合双荧光发射现象,表明D-HBF发生了ESIPT反应16。
图S1展示了N*和T*态斯托克斯位移与溶剂极性因子P(ε)和Δf之间的相关性。N*发射的斯托克斯位移与两种溶剂极性因子都显示良好的线性关系。线性拟合的斜率表明,溶剂极性对N*的斯托克斯位移有显著影响,而对T*的斯托克斯位移影响则不明显。其中,N*和T*态的相对发射强度比(IN*/IT*)随着溶剂极性的增加而逐渐变大,表明DHBF发生的ESIPT反应对溶剂的极性具有一定的依赖关系。
3.2 荧光动力学研究
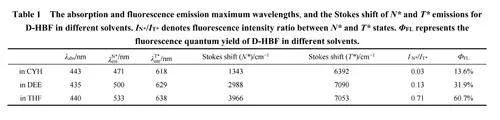
图2给出D-HBF在不同溶剂中测得的、在N*和T*特征发射波长下的荧光动力学衰减曲线(采用TCSPC技术),内插图显示了条纹相机测量得的lt;600 ps的荧光动力学。在405 nm激发下,N*态在仪器响应函数时间内生成,呈现二指数过程衰减:发生在数百皮秒内的快过程和发生在几个纳秒内的慢过程。N*的快速衰减伴随着T*快生成,表明两者之间存在动力学相关性(图2内插图所示)。当T*的生成达到最大值后,又在15 ns内衰减完全,相应的动力学分析结果总结于表2中。
动力学拟合结果表明:溶剂极性增加导致N*快衰减过程(~数百皮秒内)时间常数变长而相对占比减少,该过程对应于N*通过ESIPT转移到T*的时间常数;N*态的慢衰减过程时间常数与T*态的衰减时间常数相似(范围在2.27–3.47 ns),且随溶剂极性增加而减小。T*和N*衰减时间常数的相同表明它们之间存在快速ESIPT动态平衡9。
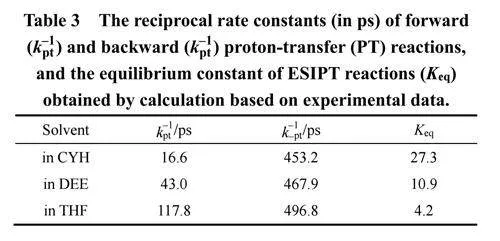
根据示意图1中所示的反应模型,可使用公式(1)和(2)来计算ESIPT的正向(kpt)和反向(k−pt)质子转移(PT)速率24,25。其中,A1和A2分别代表N*态的快速和慢速衰减物种的占比,τ1是表2中N*快速衰减物种寿命。最后,计算得到的速率常数以倒数的形式展示在表3中。
结果表明:随着溶剂极性增加,kpt和k−pt都减少,但kpt的变化比k−pt更明显。所以,ESIPT平衡常数Keq随溶剂极性增加而降低,表明ESIPT平衡向N*态移动16。
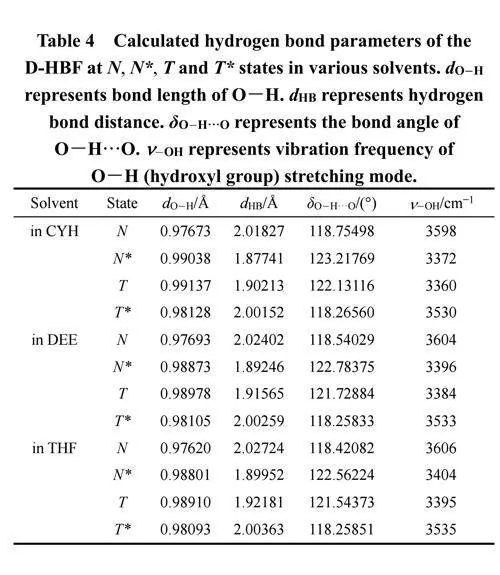
3.3.1 几何结构信息(氢键参数:长度、角度和O―H伸缩模式的IR振动频率)
4-羰基与3-羟基之间的分子内氢键强度是ESIPT发生的结构基础。本文分别计算了3-羟基的键长、键角以及红外(IR)振动频率进行验证2。使用DFT 和TD-DFT 理论结合的B3LYP-D3/6-311+g(d,p)方法,分别对D-HBF在三种溶剂中处于N、N*、T和T*态的构象进行优化,优化过程中没有对键长、键角或二面角施加任何限制。同时,获得了D-HBF在N、N*、T和T*态的振动频率且没有虚频,证实优化结构都处于局部能量极小点。
D-HBF在光激发过程中,3-OH键长(dO―H)从N态跃迁到N*态时变长而氢键距离(dHB)缩短(如表4所示)。以上结果表明,在N*态下氢键的强度增长,比N态更易发生质子转移,所以在这些溶剂中N*态存在ESIPT反应。此外,N*态下的O―HꞏꞏꞏO键角(δO―HꞏꞏꞏO)比N态更接近180°,进一步证实N*态下氢键增强。而且,从N态到N*态的转变过程中3-OH伸缩振动模式的红外振动频率向低频移动,也支持了上述观点。在计算结果中可以看到溶剂极性对分子内氢键的影响显而易见。随着溶剂极性的增加,N态下的氢键长度(dHB)从2.01827 Å增加到2.02724 Å,而在N*态下从1.87741 Å增加到1.89952Å。尽管两种态下的键长都随着溶剂极性的增加而逐渐变长,但N*态下的变化更为显著。同样,3-OH伸缩模式的红外振动频率随着溶剂极性的增加向高频移动,在N*态下频率的变化更为明显。其中,N态,从3598 cm−1增加到3606 cm−1,N*态从3372cm−1增加到3404 cm−1。
总结来说,N*态相对于N态对溶剂极性变化更为敏感。当溶剂极性增加时,激发态下的分子内氢键减弱。所以,ESIPT过程逐渐变困难。T*的溶剂极性依赖性方面与N相似,而T则与N*相似。以上结构变化最终反应在热力学上的变化,影响到系统内部的动态变化。
3.3.2 前线分子轨道分析
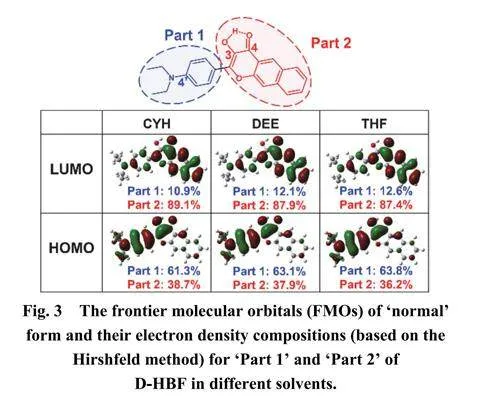
图3展示了D-HBF在CYH、DEE和THF中的前线分子轨道(FMOs)的电子密度图,包括最高占据分子轨道(HOMO,对应于N)和最低未占据分子轨道(LUMO,对应于N*)。通过对比讨论分子骨架上的电子分布以及电子转移行为。D-HBF的T和T*对应的FMOs作为参考,在Supporting Information中以图S2展示。光激发后,HOMO中的电子到跃迁到LUMO,导致电子分布不平衡进行重组。根据图3中所示的电子密度分布发现D-HBF从HOMO到LUMO的跃迁呈现π–π*特征。为了详细讨论和分析电子分布和电子转移行为,分子骨架被划分为两个部分(Part1和Part2),使用Hirshfeld方法26计算了两部分的电子密度百分比。D-HBF在三种溶剂中从HOMO到LUMO的跃迁时,Part1的电子密度从68%减少到14%,而Part2的电子密度从32%增加到86%,发生了明显的电荷转移。造成羰基上的氧原子电子密度增加,而羟基上的氧原子电子密度减少,氢键强度显著增强,为ESIPT过程的发生提供了可能性。其中,随着溶剂极性的增加,LUMO轨道上Part1上的电子强度百分比增加,Part2上的电子强度百分比减少。表明溶剂极性的增加,N*态的相应氢键强度降低,ESIPT过程变得更加困难。
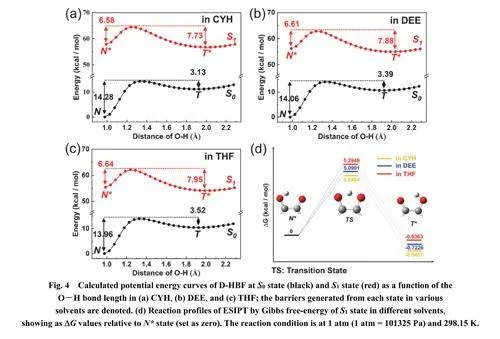
3.3.3 ESIPT反应机理分析
图4使用B3LYP-D3/6-311+g(d,p)方法计算了三种溶剂中ESIPT反应的势能曲线及其相应的势垒。在基态下,质子转移反应是吸热过程,它们正向质子转移的势垒(14.10 kcal∙mol−1 , 1 cal =4.1868 J) 远大于逆向质子转移势垒(3.35kcal∙mol−1)。相比之下,S1态下的正向ESIPT反应是放热过程,正向质子转移的势垒(~6.61 kcal∙mol−1)小于且接近逆向质子转移势垒(~7.85 kcal∙mol−1)。所以,质子转移反应倾向于S1态,由N*向T*转移。此外,溶剂极性的增加导致质子转移势垒升高,正向ESIPT反应变困难。图4d中计算得到的正向ESIPT 反应的吉布斯自由能(CYH , −0.9457kcal∙mol−1;DEE,−0.7226 kcal∙mol−1;THF,−0.6363kcal∙mol−1)随着溶剂极性增大而变大,也进一步验证了正向ESIPT反应变困难。从图4的计算结果上,溶剂极性的增加导致逆向ESIPT反应(从T*回到N*)能垒也在增加,质子转移变困难。以上计算得到的热力学结果与实验测量的动力学结果一致。
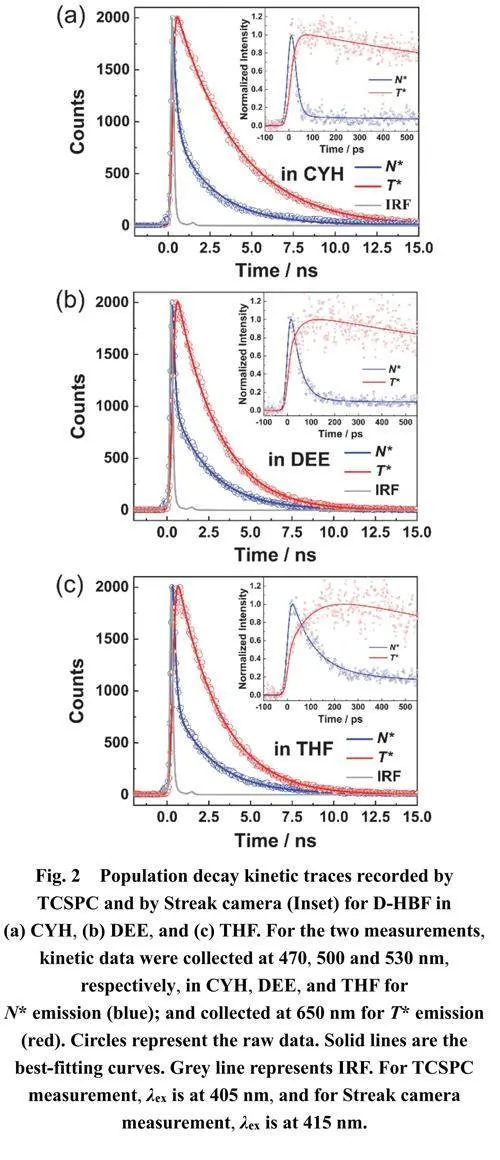
D-HBF与之前对4’-N,N-二乙氨基-3-羟基黄酮的ESIPT机制研究16结论相似,从N态激发到N*态时偶极矩存在显著差异(如表5所示)。N*态相比于N态和T*态经历溶剂稳定化作用,溶剂极性的增加导致N*态能量相比于T*态降低更显著,有利于ESIPT化学平衡向N*态移动。
综上,氢键强度是决定不同溶剂中ESIPT反应驱动力的关键因素。在第一激发态中,氢键相互作用的程度随着溶剂极性的增加而减弱,导致激发态下正向和反向质子转移的能垒都增加。其中,N*态具有ICT特征,对溶剂极性更为敏感。随着溶剂极性的增加,ESIPT反应平衡向N*态移动。最终,溶剂极性的增加导致D-HBF的双重荧光强度比IN*/IT*降低,ESIPT反应被抑制。
4 结论
本研究采用实验与理论计算相结合的方法,探讨了溶剂环境对D-HBF的ESIPT影响。研究结果表明,D-HBF具有ESIPT行为,且具有双发射峰的特征,其强度比受溶剂调控。为了进阐明ESIPT机制,我们采用了DFT和TDDFT方法结合B3LYP泛函和6-311+g(d,p)基组进行理论计算。通过比较DHBF在不同溶剂中的基态(S0)和激发态(S1)之间的分子内氢键的键长和键角,确定ESIPT反应通过激发态氢键增强机制发生。此外,溶剂极性的增加,计算得到的S1状态下O–H伸缩模式的红外吸收频率变大,也进一步证明了激发态下氢键强度再减弱。前线分子轨道(FMOs)也显示了D-HBF具有电荷转移特征,表明ESIPT通过激发态氢键增强机制发生。为了进一步确认ESIPT发生机制,分别计算了D-HBF在S0和S1态下的势能曲线。计算结果表面,质子转移过程更倾向于在S1态发生,而溶剂的极性导致ESIPT势垒升高,质子转移变困难。综上所述,溶剂极性可以调控D-HBF的双发射荧光强度比(IN*/IT*),为D-HBF作为环境极性敏感的生物荧光探针的潜在应用奠定基础。
