基于小数据集的机器学习预测酰胺键合成转化率
2025-02-20李兴海吴志森张利静陶胜洋
关键词:酰胺键合成;机器学习;特征描述符;随机森林算法;小数据集
1 引言
随着人工智能算法的快速发展,机器学习(ML)正在成为一项日益重要的数字技术1。目前,ML在多个领域得到广泛应用,特别是在自然语言处理2、医疗诊断3、人脸识别4等方面。近年来,ML在化学领域取得了显著成就,涵盖化学反应预测5、逆合成分析6、理论化学计算7和药物发现8等。例如,Doyle等人9成功训练了一个随机森林模型,用于准确预测Buchwald-Hartwig偶联反应的产率。Norquist等人10使用支持向量机模型在预测钒硒酸盐晶体制备方面取得了89%的成功率。Grzybowski等人11通过图卷积神经网络在毫秒时间尺度上准确预测了C―H酸的pKa值。此外,Jensen等人12采用神经网络模型筛选化学环境(溶剂、反应物和催化剂)和反应温度。Denmark等人13使用支持向量机和深度前馈神经网络算法预测了手性磷酸催化的硫醇与亚胺加成反应的选择性。此外,Sigman等人14提出了一种数据驱动的工作流程,用于建立预测不对称催化中对映选择性的统计模型。这些研究凸显了ML在合成化学中的关键作用。
然而,利用ML预测有机合成过程遇到了各种挑战。要获得高度准确的预测结果,通常需要大量数据集用于训练和测试,有时包括数十万甚至数百万的样本数据。例如,Waller等人5,15基于从Reaxys数据库收集的350万条反应数据训练了一个深度神经网络模型。Coley等人16使用数百万条美国专利和Reaxys数据库的反应数据来训练人工智能算法。尽管实验获得的数据能够提高准确性和可信度,但快速收集大量准确和有效的数据仍然具有挑战性。例如,默克制药公司需要使用专业的高通量反应器和众多UPLC-MS,才能在一天内完成1536个Buchwald-Hartwig偶联反应17。
在分子生物学和药物合成领域,各种高质量和低成本的高通量实验(HTE)技术在过去十年中已被广泛采用18–20。例如,在生物化学领域广泛使用的低成本聚丙烯96孔板,可以耐受各种溶剂并有助于多个反应的筛选。然而,在有限的时间内筛选数万个反应条件对研究人员来说仍然是一个重大挑战。因此,为了获得相对准确预测结果,使用有限的数据集进行ML成为了必要选择。
在本研究中,我们通过芳香胺与羧酸的反应,结合高通量实验和ML技术来预测其生成酰胺的转化率。酰胺键在众多药物化合物中普遍存在21–23,2017年新批准的药物中有60%的分子结构中含有酰胺键24。一些抗新冠药物分子,如奈玛特韦、莫诺拉韦和阿兹夫定,也都含有酰胺键25,26。然而,酰胺合成反应仍然面临着诸多挑战,包括高生产成本、有限的原子利用率以及多变的反应条件27,28。预测影响酰胺合成的关键因素具有重要意义。
本研究在确保反应的摩尔浓度和当量比等条件一致的情况下,采用96孔板进行高通量实验,以研究芳香胺和羧酸在不同偶联剂和溶剂条件下的反应结果。获得了包含1152个反应的小数据集,并成功使用随机森林算法分析出反应过程中最具影响力的因素。通过小规模地合理调整训练集,可以显著提高ML在预测未知芳香胺转化率方面的准确性,从而能够通过相对较少的实验样本预测未知反应。
2 实验部分
2.1 高通量实验采集数据
数据集的质量是影响ML算法分析准确性的关键因素。为了获取酰胺合成反应的实验数据,我们采用了高通量实验,来减少文献数据中不一致的实验当量比和不同实验环境对ML模型带来的潜在系统误差。在整个实验过程中,使用96孔聚丙烯(PP)板进行芳香胺和有机酸的酰胺合成。PP对常见的有机溶剂和化学品具有耐受性,为高通量商业反应器提供了一个经济的替代方案。这显著降低了高通量实验的成本。每个孔中可容纳约500μL的反应液,从而显著降低了试剂消耗、成本和对环境的影响。
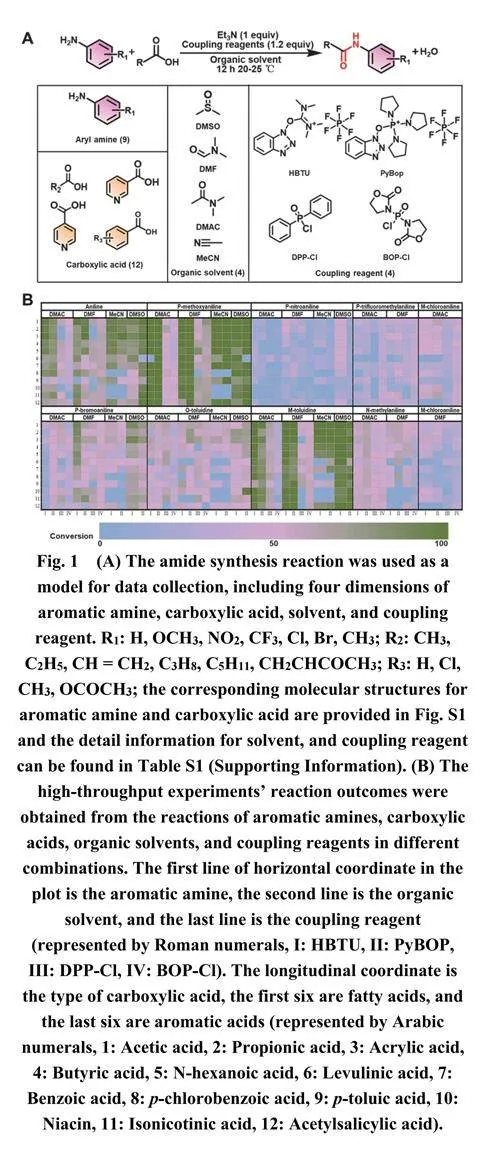
在酰胺键合成中,偶联试剂、溶剂、反应物的分子结构、酸度等多个因素都起着重要作用。在制药行业,许多酰胺化反应是在室温下进行的。因此,我们选择了活化羧酸的偶联试剂与芳香胺在室温下的反应作为ML的测试反应,以研究影响酰胺键合成的因素。如图1A所示,进行了涉及9种芳香胺、12种有机酸、4种有机溶剂、4种偶联试剂和1种碱性添加剂的微摩尔级高通量酰胺化反应。为了直观得获取酰胺合成反应的结果,采用高效液相色谱法测定芳香胺的转化率。在整个实验过程中,使用了十二个96孔板来进行1152个反应,包括1039个反应数据和113个未检测到转化的结果。反应数据在图1B中以热图形式展示,其中每个方格对应一个单独的反应条件,结果基于芳香胺的转化率绘制并以不同颜色显示,以便进行简单直观的比较。整个数据收集过程使用了11.8 g的反应物和5004 mL的溶剂,成本不到600美元(参见补充材料中的表S1)。
2.2 利用DFT提取特征描述符
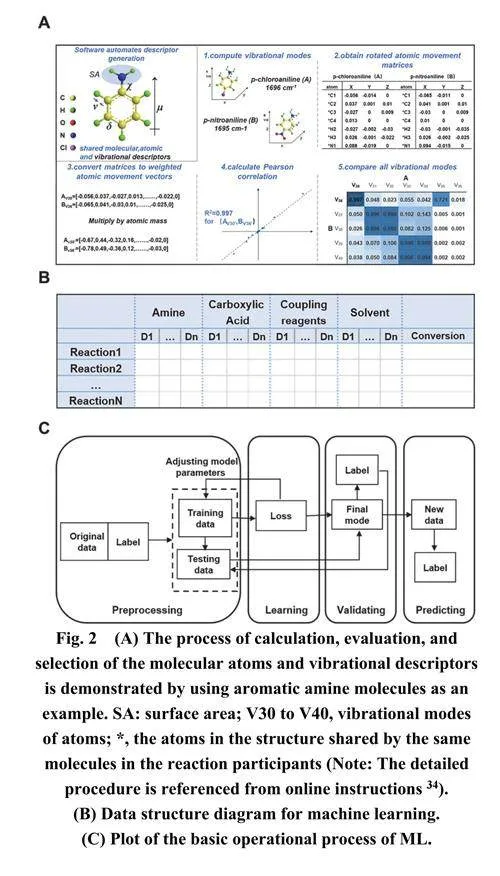
描述符是ML的基础,精确确定并选择与研究对象相关的描述符十分必要29。接下来,我们专注于选择适当的描述符。使用密度泛函理论(DFT)方法计算的分子描述符为探索化学反应机理提供了宝贵的见解。例如,Aydogdu等人30使用DFT方法计算了磺胺(SAs)的电子性质、全局描述符和局部描述符,确定了SAs与羟基自由基之间的反应机理。Ma等人31将DFT与机器学习相结合,预测铜催化的P―H插入反应中的过渡态和产率。各种量子化学计算程序产生多样的分子描述符,包括偶极矩、溶解度和轨道能级等32,33。受Doyle等人9的工作和在线教程34的启发,我们使用Spartan中的DFT、频率和性质计算,结合Python脚本程序,计算和提取了酰胺合成反应参与物的特征描述符。该软件计算了与反应相关的分子、原子和振动性质,并生成了包含描述符和转化率的建模数据表。在选择振动描述符时,我们考虑了不同原子的运动差异,将每个原子的运动乘以其原子质量,以强调重量在振动中的重要性。然后使用皮尔逊相关系数来确定需要提取和保留的振动向量。在本研究中,仅考虑R2 gt; 0.5且值大于同行同列中任何其他条目的振动模式作为匹配振动,最终被纳入建模数据表。提取的与该反应系统相关的描述符总计76个,用于表征每个反应。图2A说明了芳香胺分子的操作提取过程。补充材料中的第III部分和第IV部分的图S2、S3提供了详细的操作过程描述。分子描述符包括轨道能量(EHOMO和ELUMO)、电负性、偶极矩、椭圆度、表面积等。原子描述符包括原子的静电荷和核磁共振(NMR)位移。振动描述符包括振动频率和强度。具体而言,芳香胺有30个描述符,羧酸有21个,偶联试剂有10个,有机溶剂有15个。
2.3 数据归一化
由于描述符的多样性,每个描述符采用的评价指标不同,导致描述符量级差异较大。这种差异性在ML模型训练过程中会显著影响不同描述符的参数权重设置,较大的值往往占主导地位。因此,在训练ML模型之前,有必要对输入数据进行归一化,确保所有数据分布在[−1, 1]范围内。这种归一化使输入数据处于相同的量级,减轻了异常值的影响。在对输入数据进行归一化后,将代表转化率的输出数据纳入数据矩阵。随后剔除存在缺失值的行和列,生成用于ML模型训练的数据集,如图2B所示。
2.4 数据集划分
与先前报道的工作相比,本研究中的转化率数据是在严格调控反应条件下通过实验获得的,确保了高度的可靠性。然而,1152个反应条目的数据集是相对有限的;因此,在这些限制条件下开发一个有效的ML模型变得至关重要。对于机器学习模型训练,我们利用获得的1152个数据点(图1B)进行训练和验证。最初,我们采用标准ML模型训练方法。第一组数据被随机分为70%的训练集和30%的测试集。描述符作为输入值,而反应转化率作为输出值。矩阵的每一行对应一个反应样本,每一列代表样本的特定特征或结果标记。在ML模型的输入和输出值之间建立了映射关系,以便可以训练模型并获得最优模型。随后基于最佳模型对未知样本的反应性能进行预测。图2C说明了建立映射关系和预测未知样本的基础ML流程。
2.5 模型训练与评估
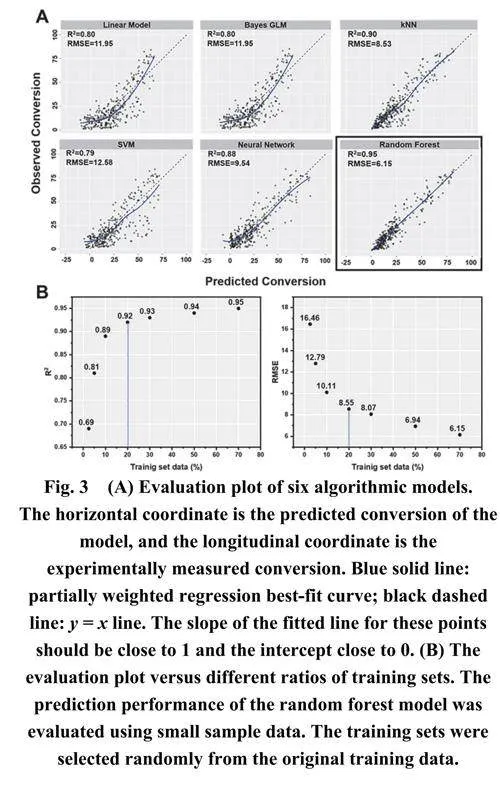
本研究使用R语言进行ML模型训练和性能评估,选择了六种算法用于ML模型训练:线性回归、k近邻(kNN)、支持向量机(SVM)、贝叶斯广义线性模型(GLM)、神经网络和随机森林。这些算法在化学合成的ML建模中经常被使用,并展示出了显著的预测能力9。在训练过程中,采用十折交叉验证方法35来提高模型的预测性能。使用决定系数(R2) 36和均方根误差(RMSE) 37来评估模型性能。通常情况下,具有高R2和低RMSE的ML模型被认为具有优秀的预测性能。经过训练后,六种算法模型对测试集展现出了不同的预测结果。
3 结果与讨论
图3A说明了线性回归、贝叶斯GLM和SVM展现出相似的预测性能,R2值约为0.8,RMSE值约为12。相比之下,神经网络和k近邻产生了更准确的预测,R2值高达0.9,RMSE值低至8.5。值得注意的是,随机森林算法展现出优越的预测性能,拥有0.95的R2值和6.15的RMSE值。与kNN相比,随机森林表现出更好的不平衡数据处理能力和更强的适应性,且不太容易过拟合。多重随机抽样方法可能促成了随机森林模型的优越性,该方法能够构建多个决策树模型。通过汇集多个低精度决策树模型的预测结果,更全面地识别了关键描述符,消除了干扰信息的影响,从而产生了具有强大泛化能力的模型。
随后, 使用不同比例的第一组反应数据(2.5%、5%、10%、20%、30%、50%、70%、80%和90%)作为训练集来训练随机森林模型。如图3B所示,分别获得了不同比例下预测性能的评估结果。随机森林模型即使在训练样本较小的情况下也表现出优秀的预测性能。与图3A中其他模型的训练结果相比,随机森林模型仅使用20%的反应数据进行训练就实现了0.92 的R2值和低至8.55 的RMSE值。这优于使用70%数据训练的线性回归、贝叶斯GLM、SVM、kNN和神经网络模型。值得注意的是,20%的数据集仅包含230个实验,表明随机森林模型可以在小规模数据集上实现可靠的预测精度。因此,随机森林模型对于常规反应优化和底物筛选是可行且有利的。
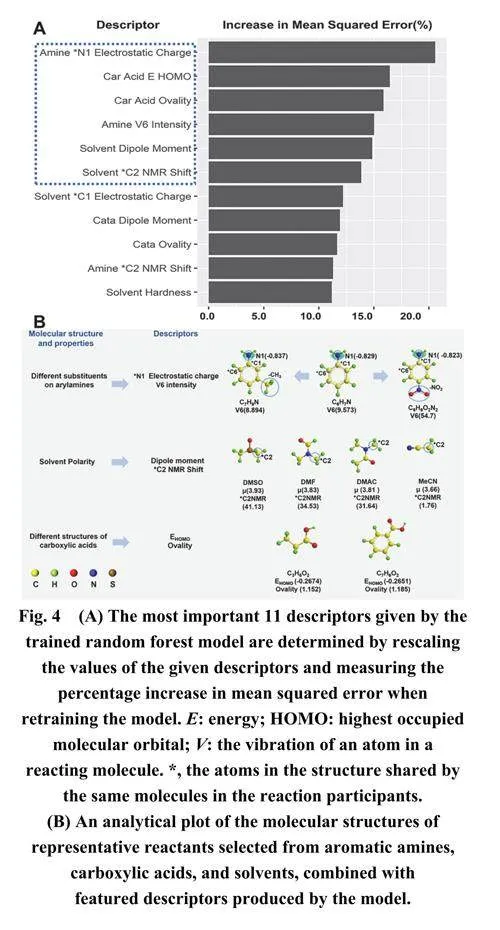
鉴于随机森林模型表现出优异的预测性能,我们使用R语言中的随机森林工具包评估了构建模型的描述符的相对重要性。该模块在训练随机森林模型后确定了特定特征描述符对预测芳香族胺转化率的相对重要性。描述符重要性通过随机扰乱描述符值并重新训练模型时模型均方误差(MSE)的增加百分比来衡量。图4A基于MSE的百分比增加展示了训练后随机森林模型的前11个关键描述符。MSE增加越大,表明该描述符在模型构建过程中越重要。
图4A表明,芳香胺、羧酸和有机溶剂的描述符对酰胺反应的转化率有较大的影响。预测反应结果的前六个重要描述符是芳香胺分子的*N1静电荷和振动强度、有机溶剂的偶极矩和*C2核磁共振位移,以及有机羧酸的EHOMO和椭圆度。先前的研究表明,芳香胺上的取代基类型和有机溶剂的极性对酰胺反应的转化率有显著影响38–41。带有给电子基团的芳香胺通常表现出比带有吸电子基团的芳香胺更高的反应活性,有利于酰胺键的形成。极性较大的有机溶剂更有利于合成酰胺键。在羧酸中,脂肪酸比芳香酸更容易形成酰胺键,这主要是因为较大的空间位阻效应不利于酰胺合成反应。
利用重要性分析的结果和我们对酰胺合成的认识,我们从芳香胺、羧酸和有机溶剂中选择了代表性的反应分子。结合这些分子与模型的描述符进行详细分析。图4B说明了芳香胺上不同取代基如何影响氨基的*N1静电荷和芳香胺分子的振动强度。六个描述符的具体数值是通过量化计算得出的。例如,含有给电子基团的间甲苯胺的*N1静电荷从−0.829增加到−0.837。根据这些数值,芳香胺分子的V6振动强度减少了0.679,这提高了底物的反应活性并促进了酰胺键的形成。结合HTE的数据发现,有机溶剂的极性(可通过偶极矩和*C2核磁共振位移推测)随着偶极矩和*C2核磁共振位移的增加而促进酰胺键的形成。诸如脂肪酸和芳香酸,不同结构的羧酸的椭圆度和EHOMO是影响酰胺键形成的因素。总体而言,该模型产生了相对准确和可靠的分析结果。
为了提高机器学习模型的可解释性,采用了沙普利可加性特征解释方法(SHAP)和累积局部效应(ALE)方法来分析所选择的特征描述符(详细结果可在图S4、S5中找到)。SHAP图表明,对酰胺键合成影响较大的特征描述符主要是芳香胺分子的*N1静电荷和振动强度,以及偶联试剂上的椭圆度和静电荷。在ALE图中,影响较大的特征描述符包括芳香胺分子的*N1静电荷、振动强度和频率、胺的EHOMO、偶联试剂的表面积和偶联试剂分子的体积。这些结果表明,这两种方法在解释特征描述符重要性方面与R语言的重要性分析相当,从而验证了R语言分析结果的可靠性。描述符的相对重要性在识别影响酰胺键合成转化率的关键因素方面证明是有用的,为筛选反应条件和讨论反应机理提供了重要指导。
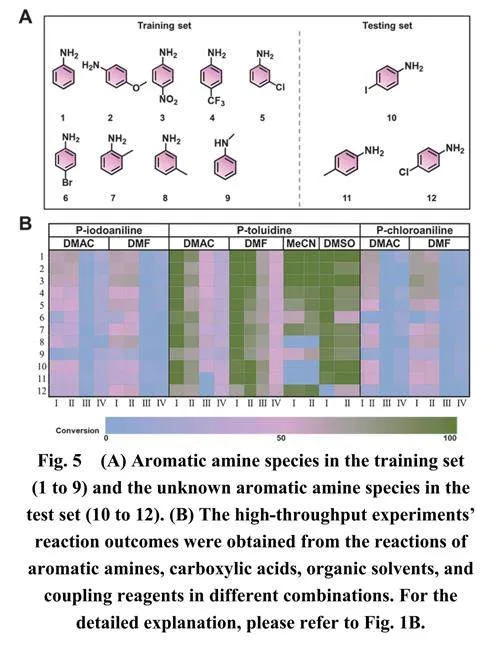
基于随机森林模型及其重要性分析结果,我们旨在使用九种已知芳香胺的反应数据来对未知芳香胺进行预测(图5)。基于结构相似性,选择了对碘苯胺、对甲苯胺和对氯苯胺作为未知芳香胺(如图5B所示)。为了获得随机森林模型的预测值对应的观测值,使用四个96孔板进行了总共336个反应,包括316个反应数据点和20个未检测到转化率的数据(如图5B所示)。
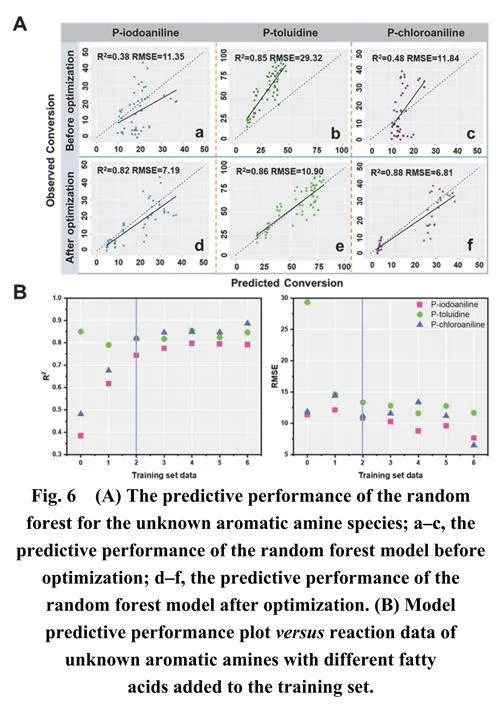
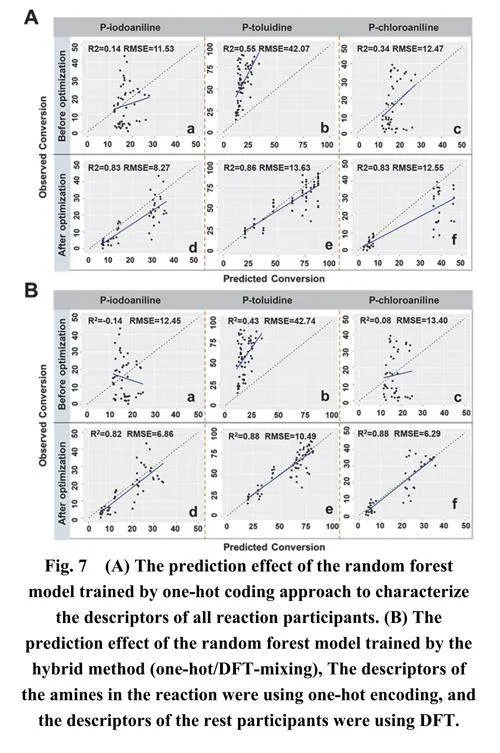
预测结果如图6A (a–c)所示。然而,随机森林模型对这三种未知芳香胺的转化率预测并未提供有效的结果。尽管模型对对甲苯胺的预测达到了0.85的高R2值,但RMSE值显著偏高,为29.32 (图6A-b)。观测值与预测值的对比图显示所有数据点都位于对角线之上,表明模型对反应转化率的预测往往低于观测结果。总的来说,这表明模型在预测反应转化率时存在显著误差。
造成这个结果的因素可能有多个:(1)与其他使用数千或数万个数据点的研究相比,用于模型训练的数据量相对有限。仅使用1152个数据点的本研究仍存在巨大的数据缺口。(2)酰胺键合成反应的原理可能并不完全遵循相似性原理;不同分子表现出不同的化学活性,即使结构相似的分子在特定反应中也可能表现不同。(3)胺化反应涉及多个参与者,影响芳香胺转化的因素又多种多样,不同因素之间可能存在协同效应。因此,分别分析单个因素对转化率的影响可能会导致一些反应信息的丢失。(4)人为因素可能影响实验过程。用于构建ML模型的描述符描述了“纯化学”信息,如反应物、偶联试剂、有机溶剂等的结构性质、电荷信息和振动频率等。ML在预测“纯化学”问题方面表现出色。然而,实验室通常会基于经验或传统,在试剂/溶剂的使用方式和典型反应条件的选择上形成一定的习惯和偏好。这些习惯和偏好有时难以量化为各种“描述符”。
在短时间内快速将实验数据增加到数万个会显著增加研究的时间和经济成本。为了应对这些挑战并在不显著增加实验数据的情况下提高模型预测准确性,我们尝试调整数据集的分布。我们将三种未知芳香胺与六种脂肪酸的反应数据添加到训练集中进行模型训练,然后使用该模型来预测它们与六种芳香族羧酸的反应。图6A (d–f)所示的结果表明,这三种芳香胺的R2值均大于0.8,平均值为0.853,RMSE显著低于之前的结果,平均值为8.3。这表明预测结果有了实质性的改进。类似地,我们尝试使用未知芳香胺与芳香族羧酸的反应数据来预测它们与脂肪酸的反应性能,这也产生了良好的预测结果,平均R2值为0.887,平均RMSE值为11.63 (图S6)。总的来说,这两种处理方法都能提高ML模型的预测准确性,表明将一些与未知分子相关的反应数据添加到训练集中对提高目标反应的预测准确性至关重要。
为了进一步证实调整训练集能提高对未知胺分子的预测准确性,采用了独热编码(one-hotencoding)方法进行模型训练和优化。使用独热编码对四种未知胺的预测评估的平均结果为R2 =0.84,RMSE = 11.48 (图7A),这一结果与使用基于DFT的分子描述符获得的结果非常相似(R2 =0.85,RMSE = 8.3)。这表明无论是使用DFT还是独热编码进行调整训练集,特别是加入未知胺的数据,都能显著提高模型的预测能力。此外,用于转化率预测的混合训练方法(one-hot/DFT-mixing)获得了平均R2 = 0.86和RMSE = 7.88 (图7B),这与完全使用基于DFT的分子描述符的性能非常接近,没有显著差异,详细结果可在图S7中找到。这些结果表明,尽管数据集仅有1152个数据点,未知胺分子的信息对模型预测结果有实质性影响。从实际角度来看,在目标反应的合成过程中,反应体系并非完全是黑箱,纳入一些已知信息(如初始原料或目标产物等)可以显著减少机器学习预测所需的数据量,这对指导有机合成反应具有实际意义。
在训练数据集方面,我们尝试在随机森林模型训练集中包含芳香胺与不同数量脂肪酸的反应数据,然后使用训练好的模型来预测三种芳香胺分子与六种芳香族羧酸的反应。如图6B所示,当添加两种或更多脂肪酸时,改善效果相对更为显著。对于添加到训练集中的两种脂肪酸,共15种组合,通过将未知芳香胺分子与两种脂肪酸的反应数据纳入训练集,观察到R2显著提升和RMSE的降低(具体信息可在表S2中找到)。随着训练集中数据量的增加,R2逐渐提高,RMSE逐渐降低,随机森林模型的预测性能稳步提升。尽管基于小样本数据集的随机森林模型在预测未知反应的转化率方面存在局限性,但通过添加少量相关分子反应数据,其性能可以显著提升,使其达到可接受范围。这种方法仅需要二到三种简单反应物就能产生数据集,对于预测大量未知反应具有重要意义,可以显著减少实验工作量,加快反应研究的进展。
4 结论
通过构建包含1152个数据点的数据集,证明了基于机器学习预测酰胺键合成中反应转化率的可行性。采用六种不同的机器学习算法进行模型训练,其中随机森林算法表现出最优异的预测性能,R2值超过0.95。通过适当调整训练集数据,实现对未知芳香胺反应转化率的有效预测。对比使用不同分子描述符(如DFT和独热编码)训练的模型分析表明,在少于2000个数据的小数据集条件下,调整训练集组成能有效提升机器学习的预测性能。这为机器学习在关键分子合成领域的广泛应用提供了一种新策略。
