基于SSR标记的皱皮木瓜遗传多样性分析及品种分子身份证构建
2025-01-26李慧侯立娜王天琪毕宁宁李圣波刘忠华







摘要:【目的】皱皮木瓜(Chaenomeles speciosa)具有较高的观赏价值食用价值与药用价值,我国是皱皮木瓜的起源分布中心,研究皱皮木瓜种质资源遗传多样性,并构建品种分子身份证,以解决近年来因缺乏品种间统一的分类标准,同名异种、同种异名和品种间来源及演化不清等问题。【方法】以收集的168 份皱皮木瓜种质资源为材料,利用SSR标记结合毛细管电泳法对木瓜遗传多样性和群体内遗传分化程度进行分析,根据观测等位基因数(Na)、Shannon’s信息指数(I)和多态性信息含量(PIC)的筛选能区分全部种质的引物组合,并基于字符串编码构建DNA分子身份证。【结果】26 对引物在168 个皱皮木瓜品种中共扩增出304 个等位基因,平均每个位点扩增出11.577 个;期望杂合度(He)、I和PIC的平均值分别为0.748、1.731和0607。根据聚类分析的结果可将该群体分为2 大类,进一步分为6 个类群;种群结构分析将供试材料分为2 个亚类。从26 对引物中筛选出11 对核心引物构建了168 份皱皮木瓜种质资源的条形码和二维码身份证。【结论】所选扩增位点的变异程度高、鉴别力度大,在进行遗传多样性分析、核心引物筛选,以及指纹图谱构建中可优先选用。该研究为皱皮木瓜的良种鉴定、遗传资源管理及种质资源数据库的构建等提供了参考。
关键词:皱皮木瓜;遗传多样性;SSR;分子身份证
中图分类号:S793.9;S567.9""""" 文献标志码:A
开放科学(资源服务)标识码(OSID):
文章编号:1000-2006(2025)01-0059-10
The genetic diversity analysis and molecular ID establishment of Chaenomeles speciosa"" based on SSR markers
LI Hui "HOU Lina "WANG Tianqi "BI Ningning "LI Shengbo3, LIU Zhonghua 2
(1. National Engineering Laboratory for Tree Breeding, Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental, Plants of Ministry of Education," Key Open Laboratory of Flower Breeding and Bioengineering, National" Forestry and Grassland" Administration, Beijing Forestry University, Beijing 100083, China; 2. College of Biological Sciences and Biotechnology, Beijing Forestry University, Beijing" 100083, China; 3. Shandong Yate Eco-tech, Co., Ltd., Junan 26607 ""China)
Abstract: 【Objective】Chaenomeles speciosa has high ornamental, edible, and medicinal values. China is the origin and distribution center of C. speciosa. The genetic diversity of Chaenomeles speciosa germplasm resources was studied, and the molecular identity card of varieties was constructed to solve the problems of lack of unified classification criteria among varieties, homonymous and synoonymous, and unclear origin and evolution among varieties in recent years.【Method】A total of 168 C. speciosa varieties and SSR (simple sequence repeats) markers were combined with capillary electrophoresis to analyze the genetic diversity and the degree of genetic differentiation. The observed number of alleles (Na), Shannon’s information index (I), and the polymorphism information content (PIC) were employed to screen primer combinations that can distinguish the entire germplasm and construct DNA molecular IDs based on string codes.【Result】The results showed that 26 pairs of primers amplified 304 alleles in 168 C. speciosa varieties, with an average of 11.577 per locus. The average expected heterozygosity (He), I" and PIC were determined as 0.748, 1.731" and 0.607, respectively. Based on the cluster analysis, the population could be divided into two groups, and further into six groups. Moreover, based on population structure analysis, the tested materials was divided into two subgroups. Four pairs of core primers were selected from 26 pairs of primers to construct barcode and two-dimensional code identification cards of C. speciosa varieties.【Conclusion】The selected amplification loci have a high degree of variation and strong discrimination, and are thus suitable for applications in genetic diversity analysis, core primer screening, and fingerprint construction. The results can provide a reference for the variety identification, genetic resource management, and the construction of a germplasm resource database of C. speciosa.
Keywords:Chaenomeles speciosa;genetic diversity;SSR;molecular ID
皱皮木瓜 (Chaenomeles speciosa) 又名贴梗海棠、木瓜、贴梗木瓜,属于蔷薇科木瓜属落叶灌木。中国作为皱皮木瓜的起源分布中心,东至辽东、浙江,西至新疆、西藏,南至云南、广西,北至山西、甘肃等地均有其分布。南方的皱皮木瓜资源以宣木瓜、酸木瓜、淳木瓜和川木瓜(地方俗名)为主,果实偏小,多野生,多供观赏及药用;北方则以河南木瓜或山东木瓜为主,果实大,多为栽培品种,除观赏外,具有很高的药用及食用价值。山东是皱皮木瓜的道地产区之一,近数十年在品种选育及引种驯化等方面取得显著进展,皱皮木瓜的种植已成为传统农业的一部分,不断满足医疗和食品工业日益增长的需求[1]。皱皮木瓜具有高度可塑性表型,容易进行属内的种间杂交,杂交种具有较高的园艺利用价值,但给种质资源的鉴定和分类带来一定困难。由于品种间缺乏统一的分类标准,从而导致同名异种、同种异名和品种间来源及演化不清等问题的出现[2]。过去人们根据表型特征来评价遗传资源,但环境因素的多变性影响了表型鉴定稳定性和准确性;再者传统方法鉴定周期过长,且部分品种的果实外观非常相似,给资源保护和产品深度开发带来了极大的困难。建立一套高效、准确的品种识别方法是目前迫切需要解决的问题。
近年来,分子标记已被逐渐应用于皱皮木瓜种质资源鉴定和遗传多样性分析等领域。王明明等[3]利用22 个SRAP引物组合对27 份品种和5 份野生木瓜属种质进行聚类分析、主坐标分析及遗传多样性评价,发现C.×superba与皱皮木瓜亲缘关系较近, 可作为皱皮木瓜种下的品种群。He等[4]使用8 个AFLP遗传标记将52 个皱皮木瓜种质分为7 个类群,遗传多样性及聚类分析的结果揭示种群内遗传变异性具有较高水平,为后期的育种改良提供了基础。张艳艳等[5]运用苹果属的EST-SSR标记对木瓜属品种进行聚类分析,证明了苹果属的EST-SSR标记可用于木瓜属的遗传关系研究和资源评价。蒋小刚等[6]利用10 个ISSR引物将12 份来自全国皱皮木瓜主产区的种质资源分为3组,揭示了其有较高的遗传多样性。在众多的分子标记技术中,SSR标记因其共显性、特异性和高重复性比RAPD和AFLP等分子标记更具优势[7],而被国际植物新品种保护联盟 (UPOV) 纳为适合构建品种分子标记数据库的技术之一[8]。DNA分子身份证是在指纹图谱基础上,将图谱以字符串的形式表示,使每个品种拥有特定的数字化代码。相比于构建指纹图谱,分子身份证能够更加直观便捷地显示品种间的差异,并可通过计算机自动比对差异,避免人工比对的低效、繁琐,可在大规模品种比对中广泛使用[9],但目前鲜见关于皱皮木瓜核心引物筛选和分子身份证构建的相关报道。
本研究运用荧光标记SSR结合毛细管电泳技术,对收集到的168 份皱皮木瓜种质资源进行遗传多样性分析和群体结构分析,并通过最佳引物组合构建DNA分子身份证,以期为皱皮木瓜品种鉴定和资源保护提供参考。
1 材料与方法
1.1 供试材料
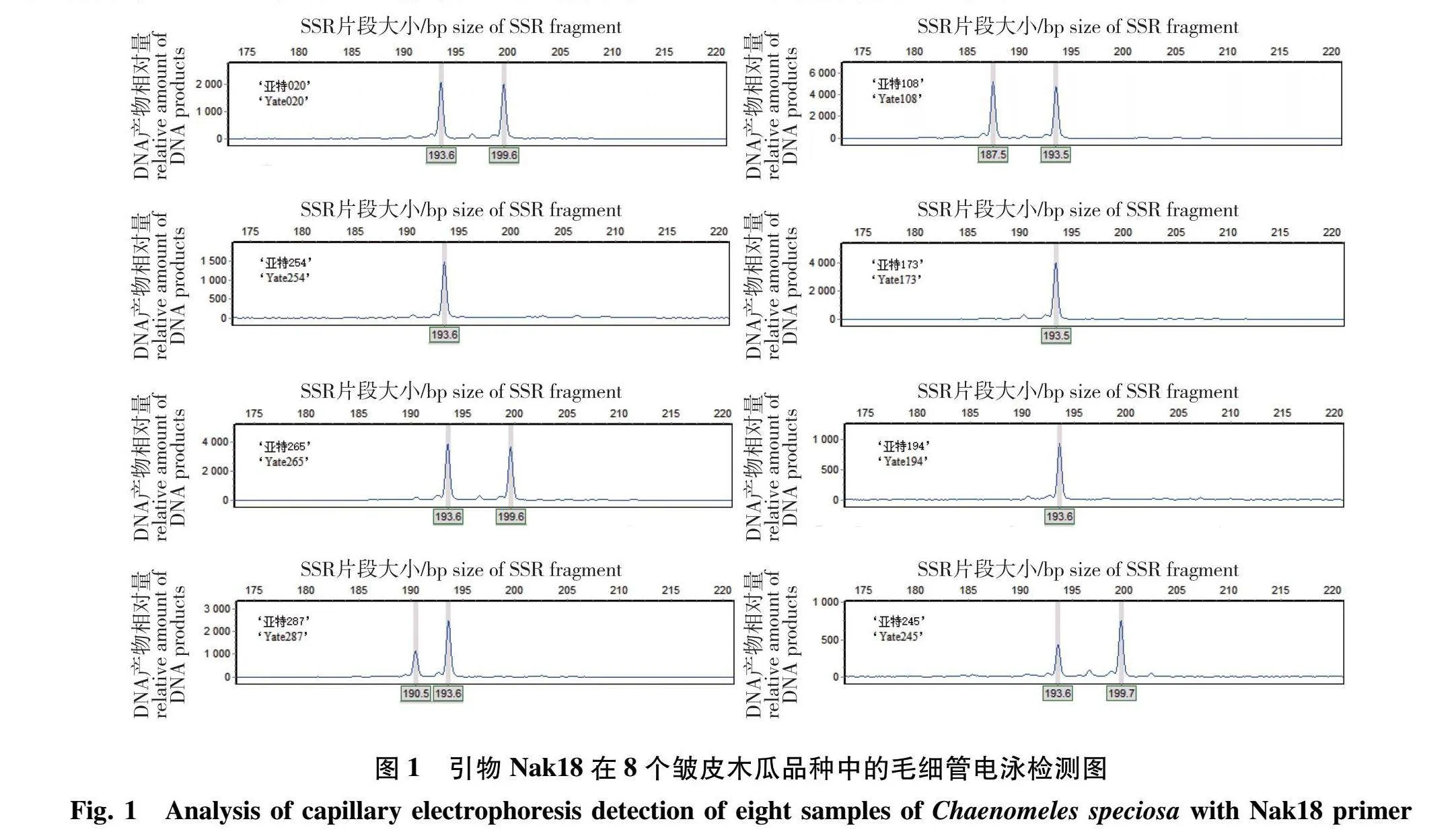
在供试168 份皱皮木瓜样品中,160 份采自山东省临沂市亚特药用植物生态园 (皱皮木瓜种质资源库),其中11 份为临沂本地的老品种,且已通过山东省林木良种审定,149 份收集自云南大理、贵州正安和湖北恩施等地,大部分也是本土品种,小部分是从我国各地野生资源中选育出的新基因型;其他8 份是从北京市周边、贵州贵阳和江苏苏州等地收集得来的传统品种,木瓜样品详细信息见附表1(https://nldxb.njfu.edu.cn)。采集各植株新生枝上完全展开的幼嫩叶片,变色硅胶干燥,备用。其中‘红香玉木瓜’(‘Hongxiangyu’)用于简化基因组测序,随机选择的12 份样品[‘红香玉’‘绿香玉’(‘Lü Xiangyu’)、‘金宝罗青101’(‘Jinbaoluoqing 101’)、‘金宝罗青106’(‘Jinbaoluoqing 106’)、‘亚特020’(‘Yate 020’)、‘亚特108’(‘Yate 108’)、‘亚特254’(‘Yate 254’)、‘亚特173’(‘Yate 173’)、‘亚特265’(‘Yate 265’)、‘亚特194’(‘Yate 194’)、‘亚特287’(‘Yate 287’)和‘亚特245’(‘Yate 245’)] DNA作为模板进行TP-M13-SSR PCR,用于筛选在不同皱皮木瓜样本间具有较高多态性的引物。
1.2 DNA提取
利用自动磨样机结合高效植物基因组DNA提取试剂盒 [天根生化科技(北京)有限公司,DP350]提取植物叶片基因组DNA。经1%(质量分数)的琼脂糖凝胶电泳检测DNA的质量,利用NanoDrop 2000分光光度计 (Thermo Scientific,美国) 检测其浓度和λ260//λ280值,将DNA样品的浓度稀释至20 ng/mL,-20" ℃保存备用。
1.3 引物筛选与PCR扩增
基因组测序产生reads,对reads进行组装产生长片段Contigs,确定Contig的方向和顺序,组装产生更长的片段Scaffolds,最后再组装连接Scaffold得到完整的染色体序列。在无参考基因组的情况下,根据北京源宜基因科技股份有限公司完成的皱皮木瓜简化基因组测序结果,利用MISA识别Contig上SSR序列。利用Primer 3 (2.3.7,http://primer3.source-forge.net/) 设计引物。根据毛细管电泳峰图的带型分布情况分析引物多态性,筛选引物位点[10-11]:① 随机挑选12 个皱皮木瓜品种用于引物初筛,通过查看已获得的所有引物毛细管电泳峰图,将峰型良好、扩增谱带数≥3、谱带间差值≥1 bp的引物用作候选引物; ② 随机选择3 个以上皱皮木瓜品种对候选引物进行重复试验和复筛,以确保候选引物扩增的稳定性。最终确定26 对带型稳定、多态性高的引物用于后续试验。
本研究共需3 种引物[12]。带有M13序列 (5′-TGTAAAACGACGGCCAGT-3′) 的上游引物,下游引物及具有荧光染料标记的M13引物 (FAM、HEX、TAMRA、ROX),均由北京睿博兴科生物技术有限公司合成。20 mL反应体系包括:2 mL模板DNA (20 ng/mL),10 mL 2×Taq Master Mix (擎科生物公司),0.08 mL (10 mmol/L) 上游引物,0.32 mL (10 mmol/L)下游引物,0.4 mL M13引物和7.2 mL的ddH2O。PCR扩增程序为94" ℃预变性3 min;94" ℃变性30 s,适宜的退火温度退火30 s,72" ℃延伸1 min,34个循环;72" ℃延伸5 min,4" ℃保存。
1.4 毛细管电泳检测及数据分析
PCR产物委托北京睿博兴科生物技术有限公司在ABI-3730XL基因分析仪 (Applied Biosystems,Foster City,CA,美国) 上进行毛细管电泳检测。应用GeneMarker 2.2.0" (SoftGenetics,美国) 软件[13]对扩增片段长度进行整理分析;利用CONVERT 1.31进行数据格式转换,利用POPGEN" 1.32[14]和CERVUS"" 3.0软件[15]计算多态性位点的等位基因数 (Na)、有效等位基因数 (Ne)、期望杂合度 (He)、Shannon’s信息指数 (I)、多态性信息含量 (PIC) 等信息;利用NTSYSpc 2.10e软件[16]计算品种间遗传相似性系数;应用PowerMarker 3.25[17]、MEGA7构建UPGMA系统树并进行聚类分析,在线软件iTOL (https://itol.embl.de/) 用于系统树优化;Structure 2.3.4软件[18]用于群体结构分析,以最大似然值原则,通过ΔK确定最佳K值,参数设置为Burn-in和MCMC (Markov chain monte carlo) 值均为10 000;K取1~10,重复10次;其余设置为默认值。
1.5 DNA分子身份证构建
采用数字加字母组合的方式构建皱皮木瓜DNA分子身份证[19],将每对引物扩增出的基因型 (带型),按分子量大小用自然数1~9进行赋值,对于9以上的基因型则用字母A—Z表示,未扩增出等位变异片段的赋值为“0”,每个带型占1位,构建字符串形式的分子身份证编码,将分子身份证编码导入条形码生成器 (http://www.t-x-m.com/) 构建条形码身份证,将皱皮木瓜品种来源、花果特征、生长特性以及字符串编码等信息导入二维码生成器 (https://cli.im/),生成可供扫描的二维码身份证。
2 结果与分析
2.1 TP-M13-SSR标记多态性分析
荧光毛细管电泳检测的结果(图1)显示,26 对引物在绝大部分皱皮木瓜品种中能获得稳定清晰的扩增片段,详细结果如表1所示。26 对引物共扩增出304 个等位基因,每个位点的等位基因数为4 (Nak26)~18 (Nak10) 个,平均每个位点扩增出11.577 个;有效等位基因数为2.061 (Nak4)~6766 (Nak15),平均为4.376;期望杂合度为0.516~0.855,平均为0.748;不同位点的PIC变化范围较大,为0.287~0.81 平均PIC为0.607。根据Botstein等[20]提出的多态信息含量标准,高多态性引物 (PIC gt; 0.5) 有20 对,中多态性引物 (05 gt; PIC gt; 0.25) 6 对。上述参数表明供试皱皮木瓜品种间具有较为丰富的多样性,所选用的26 对引物具有高度多态性。
2.2 皱皮木瓜品种亲缘关系聚类分析
利用 NTSYSpc 2.10e 软件计算出皱皮木瓜品种间遗传相似性系数 (genetic similarity coefficient,GS) 的变幅为0.029 (‘亚特149’和‘亚特201’)~0971 (‘苏02’和‘亚特028’),平均为0540。结合Excel对相似性系数矩阵分析,共得到14 027 个GS,以0.03为组距构建皱皮木瓜遗传相似性系数频率分布图 (图2a),供试皱皮木瓜品种GS集中分布在0.449~0.599之间,其中0.509~0.539的分布频率最高,占比达9.22%;GS为0.509~0989的占比5989%,表明供试皱皮木瓜品种间遗传差异较小,亲缘关系较近。
聚类结果可将168 个皱皮木瓜品种分为2 大类,即‘金宝罗青102’和‘亚特245’聚为1 类,其余的品种聚为1 类。本研究在此基础上进一步将其细分为6 个类群 (图2b)。第1 类群包括来自苏州的2个品种;第2类群5 个品种,包括采自北京的3 个品种和2 个采自山东皱皮木瓜种质库的选育品种;第3 类群、第4 类群及第5 类群分别由2 个、25 个和63 个品种组成;第6 类群是最大的一个类群,共69 个品种,含4 个采自山东的品种和3 个采自贵州的品种。此外,有2 个品种没有聚类到任何主要群体中,表明这2 个种比其他种具有更高的遗传差异,在后期的核心种质库的构建中可优先考虑。
2.3 皱皮木瓜种质遗传结构分析
通过Structure 2.3.4对供试皱皮木瓜种质进行遗传结构分析的结果显示,平均后验概率值[ln P(D)]在群体亚群数(K)从1~10的过程中呈上升趋势,并无明显拐点,因此根据Evanno等[21]的方法进一步确定亚群数。ΔK最大值出现在K= 2时,据此结果将168份皱皮木瓜材料分为2个亚类 (图3),第1亚类包括‘亚特020’—‘亚特058’,共135 个品种,第2亚类包括‘亚特197’ —‘亚特116’,共计33 个品种,两个亚群间亲缘关系稍远,亚群内不同品种间亲缘关系较高,但也存在着基因频率的差异。此外,与UPGMA聚类法对比发现,虽两种方法均可将供试材料分为2 类,但组分不同,可能是由两种方法的分类依据不同造成的。
2.4 最佳引物组合的筛选
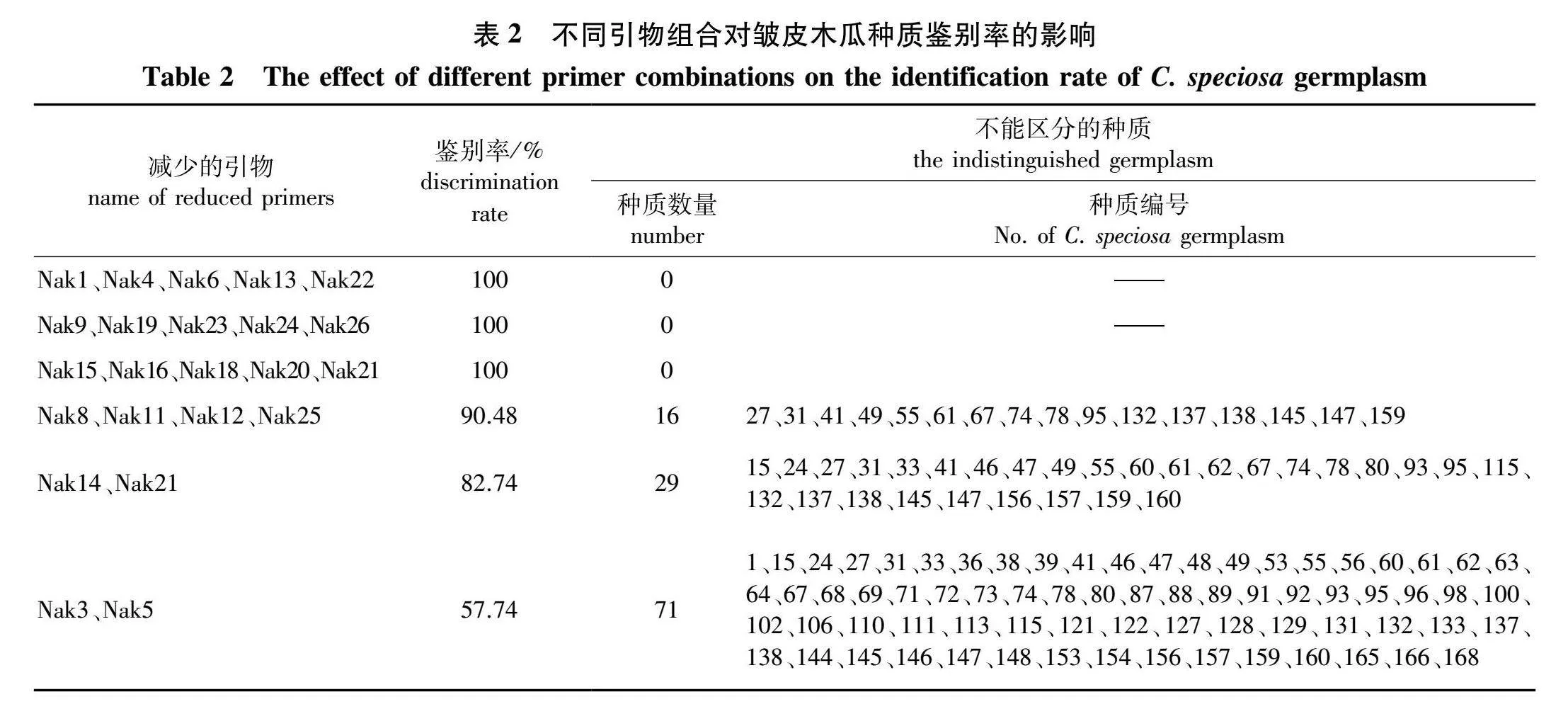
遗传多样性指数 (H)、观测等位基因数 (Na) 和多态信息含量 (PIC) 是衡量群体多样性的重要指标。如果表现出较高的H(>0.5)、PIC(>0.5) 和较多Na(≥5),说明该位点遗传变异程度高,选择余地大,可利用这样的位点进行标记辅助选择[22-23]。本着以最少的引物鉴别最多的种质的原则,利用Na、PIC值及H等指标,逐步去除参数值低的引物,通过观察引物删除对种质区分率的影响来确定最佳引物,当引物鉴别率明显下降时,留下的引物即为最佳引物组合(表2)。
由表2可知,首先去除PIC较低的5 对引物Nak1、Nak4、Nak6、Nak13和Nak2 去除后所有引物均能被区分。接着删除Na和H较低的5 对引物,去掉后并未增加不能区分的品种。以此类推,当删除H和Na较低的2 对引物时,有71 个品种不能区分,此时的引物鉴别率为57.74%,与上一步的结果 (82.74%) 相比鉴别率有明显的下降,由此可见若继续删减引物,将有大量的种质资源无法区分。综上所述,将能全部区分供试皱皮木瓜品种的11 对引物 (Nak2、Nak3、Nak5、Nak7、Nak8、Nak10、Nak11、Nak12、Nak14、Nak17和Nak25) 作为最佳引物组合,用于供试木瓜品种的分子身份证构建。
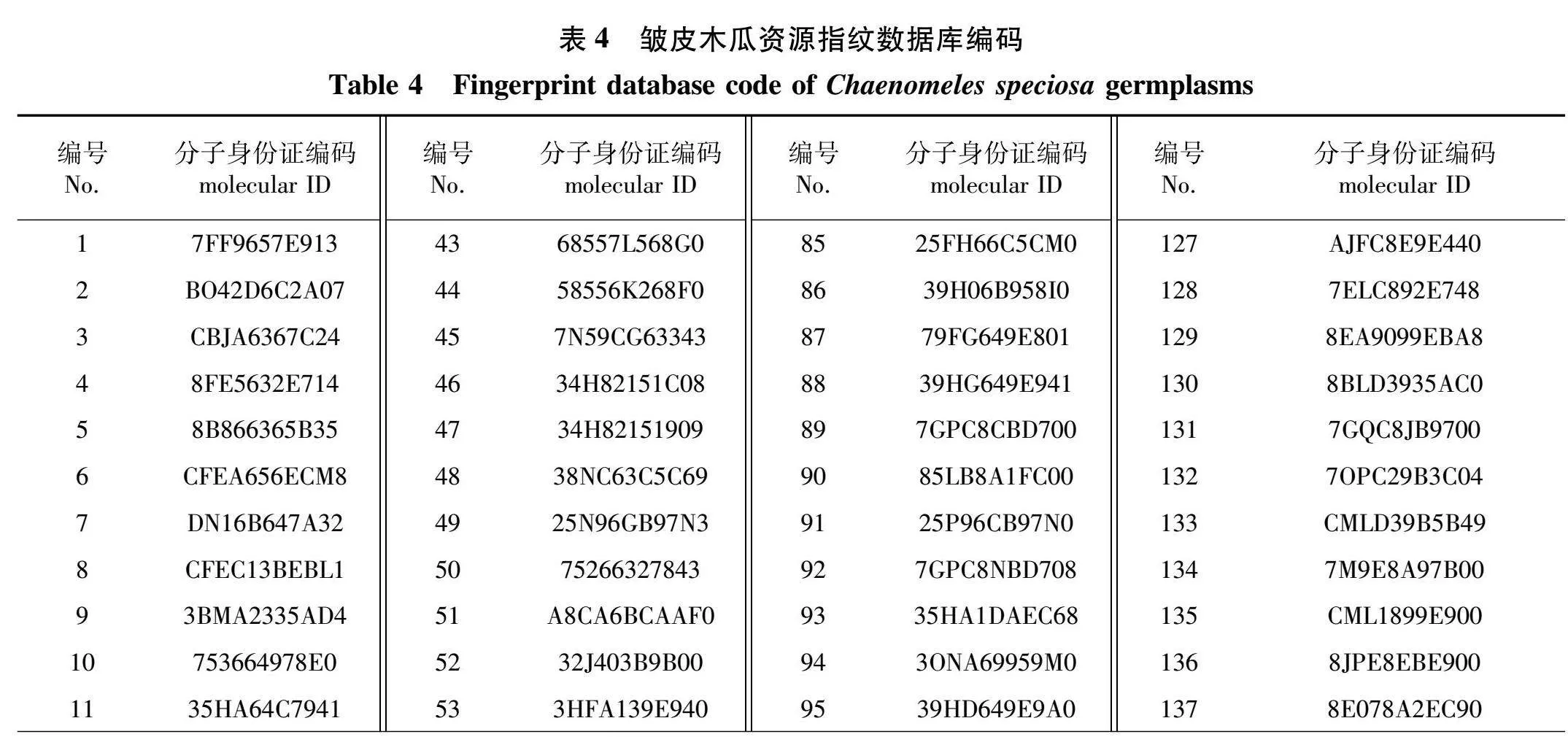
2.5 皱皮木瓜DNA分子身份证编码
利用所筛选的11 对引物 (Nak2、Nak3、Nak5、Nak7、Nak8、Nak10、Nak11、Nak12、Nak14、Nak17和Nak25),采用数字加字母组合法对168份皱皮木瓜品种进行赋值 (表3),并构建DNA分子身份证 (表4),再利用条形码生成器生成条形码DNA身份证。将皱皮木瓜相关信息导入二维码生成器,生成二维码DNA分子身份证。皱皮木瓜的条形码和二维码身份证实例见图4。
3 讨 论
随着高通量测序技术的飞速发展,人们开始利用全基因组数据开发SSR标记并建立SSR检索数据库[24-25]。但由于皱皮木瓜的全基因组数据库目前尚未建立,因此本研究将简化基因组测序技术和TP-M13-SSR PCR用于皱皮木瓜的遗传多样性分析、群体结构分析和指纹图谱及分子身份证构建,弥补了皱皮木瓜在SSR分子标记以及品种鉴定等方面的匮乏。遗传多样性分析的结果显示,本研究中的26 对引物平均Shannon’s信息指数 (I) 为1731、期望杂合度(He)的平均值为0748,表明各位点的遗传多样性存在较大差异。所选用的26 对SSR引物中多态性高的引物 (PICgt;05) 有20 对,高多态性引物占据所选引物的7692%,表明所选扩增位点的变异程度高、鉴别力度大,在进行遗传多样性分析、品种鉴定、核心引物筛选,以及指纹图谱构建中可优先选用。
在群体结构分析中,采用了UPGMA聚类和Structure软件对全部的皱皮木瓜品种进行聚类分析。全部供试皱皮木瓜种质在UPGMA聚类图中分为了2 个亚类,进一步细化又可分为6 组。基于Structure软件对其进行聚类分析也可将全部种质分为2 个亚类,但两种方法分成的两个亚类的种质组成是不同的,出现这种情况的原因可能是由于两种聚类分析的分类依据不同。有研究表明,基于数学模型的群体遗传结构分类比基于材料间的遗传距离的聚类分析更加精确[26]。也就是说,基于Structure软件聚类分析相较于基于UPGMA聚类分析有更高的准确性。因此本研究利用Structure软件划分供试材料的结果,进行后续相关分析。
目前有3种方法用于SSR毛细管电泳结果赋值编码,分别是“0/1”法、等位基因编码法、基因型编码法。“0/1”法中有效扩增片段的有无用数字“1”和“0”表示,形成字符串的赋值编码[27],但这种方法形成的字符串不够精准,且容易过于冗长,以致读取不便,因而更多地用于基于聚丙烯酰胺凝胶电泳法的指纹图谱构建。等位基因编码法按每对引物扩增出的等位基因分子量大小进行赋值编码[28]。该法对于纯合谱带多的物种影响不大,但对于区分杂合带型和纯合带型编码相同的品种存在一定困难[29]。基因型编码法按照引物的特定顺序,对扩增出的基因型进行赋值编码。该法方便统计,并能真实有效地反映引物的扩增结果,适用于较大规模的指纹图谱构建。通过分析比较3种赋值编码方式的优缺点,本研究采用基因型编码法对皱皮木瓜进行指纹图谱构建。
研究表明在用于种质资源鉴定的DNA分子标记中,AFLP因其多态性远高于RFLP和RAPD,被认为是DNA指纹图谱技术中多态性最为丰富的一项技术[9],也是目前皱皮木瓜品种鉴定研究中使用率最高的一种方法。但用于构建DNA分子身份证的标记应具有操作简便、重复性好且具有代表性等特征。而SSR标记因在不同品种间每个特定大小的等位基因序列一致而具有更高的重复性,同时SSR不需要酶切模板,操作更加简单,因而被认定为品种鉴定的首选标记。然而对于全基因组序列未公开的物种,虽然可使用简化基因组或转录组测序技术获得多态性引物,但无法保证这些引物是否均匀分布于各条染色体上。为解决上述问题,后续研究可采用多种分子标记相结合的方式弥补单一方式所造成的不足。
参考文献(reference):
[1]陈红,王关祥,郑林,等.木瓜属(贴梗海棠)品种分类的研究历史与现状[J].山东林业科技,2006,36(5):70-7 78.CHEN H,WANG G X,ZHENG L,et al.The studying history and current status of Chaenomeles[J].J Shandong For Sci Technol,2006,36(5):70-7 78.DOI: 10.3969/j.issn.1002-2724.2006.05.031.
[2]SINGH R B,SINGH B,SINGH R K.Evaluation of genetic diversity in Saccharum species clones and commercial varieties employing molecular (SSR) and physiological markers[J].Ind Jour Plant Gene Resour,2018,31(1):17.DOI: 10.5958/0976-1926.2018.00003.7.
[3]王明明,陈化榜,王建华,等.木瓜属品种亲缘关系的SRAP分析[J].中国农业科学,2010,43(3):542-551.WANG M M,CHEN H B,WANG J H,et al.Genetic relationship of Chaenomeles cultivars revealed by SRAP analysis[J].Sci Agric Sin,2010,43(3):542-551.DOI: 10.3864/j.issn.0578-1752.2010.03.014.
[4]HE J S,FAN J W,LI S B,et al.Genetic variability of cultivated Chaenomeles speciosa (Sweet) Nakai based on AFLP analysis[J].Biochem Syst Ecol,2014,57:445-450.DOI: 10.1016/j.bse.2014.09.022.
[5]张艳艳,齐红,郭庆梅,等.利用苹果EST-SSR分析木瓜属种质遗传多样性[J].生物技术通报,2016,32(7):93-98.ZHANG Y Y,QI H,GUO Q M,et al.Analysis of genetic diversity in Chaenomeles using apple EST-SSRs[J].Biotechnol Bull,2016,32(7):93-98.DOI: 10.13560/j.cnki.biotech.bull.1985.2016.07.014.
[6]蒋小刚,林先明,张美德,等.基于ISSR分子标记的皱皮木瓜遗传多样性分析[J].分子植物育种,2020,18(21):7239-7245.JIANG X G,LIN X M,ZHANG M D,et al.Genetic diversity analysis of Chaenomeles speciosa(sweet) nakai based on ISSR molecular markers[J].Mol Plant Breed,2020,18(21):7239-7245.DOI: 10.13271/j.mpb.018.007239.
[7]HECKENBERGER M,VAN DER VOORT J R,PELEMAN J,et al.Variation of DNA fingerprints among accessions within maize inbred lines and implications for identification of essentially derived varieties:II.genetic and technical sources of variation in AFLP data and comparison with SSR data[J].Mol Breed,2003,12(2):97-106.DOI: 10.1023/A:1026040007166.
[8]王凤格,赵久然,田红丽,等.农作物品种DNA指纹库构建研究进展[J].分子植物育种,2015,13(9):2118-2126.WANG F G,ZHAO J R,TIAN H L,et al.The progress of the crop varieties DNA fingerprint database construction[J].Mol Plant Breed,2015,13(9):2118-2126.DOI: 10.13271/j.mpb.013.002118.
[9]徐雷锋,葛亮,袁素霞,等.利用荧光标记SSR构建百合种质资源分子身份证[J].园艺学报,2014,41(10):2055-2064.XU L F,GE L,YUAN S X,et al.Using the fluorescent labeled SSR markers to establish molecular identity of lily germplasms[J].Acta Hortic Sin,2014,41(10):2055-2064.DOI: 10.16420/j.issn.0513-353x.2014.10.012.
[10]游倩.甘蔗种质资源的SSR遗传多样性分析及指纹数据库构建[D].福州:福建农林大学,2014.YOU Q.Genetic diversity analysis and database construction of DNA fingerprintings in sugarcane based on SSR fluorescence markers[D].Fuzhou:Fujian Agriculture and Forestry University,2014.
[11]赵久然, 王凤格. 玉米品种指纹鉴定技术研究与应用 [M]. 北京:中国农业科学技术出版社,2014.ZHAO J R, WANG F G. Research and application of fingerprint identification technology of corn varieties [M]. Beijing: China Agricultural Science and Technology Press,2014.
[12]SCHUELKE M.An economic method for the fluorescent labeling of PCR fragments[J].Nat Biotechnol,2000,18(2):233-234.DOI: 10.1038/72708.
[13]HULCE D, LI X, SNYDERLEIBY T, et al.GeneMarker genotyping software: tools to increase the statistical power of DNA fragment analysis[J].J Biomol Tech,201 22(Sl):35-36.
[14]YEH F C, YANG R C, BOYLE T. POPGENE Version 1.32: microsoft Windows-based freeware for populations genetic analysis [EB/OL]. Edmonton: University of Alberta. (1999).[2022-03-20]. http://sites.ualberta.ca/~fyeh/popgene_download.html.
[15]MARSHALL T C,SLATE J,KRUUK L E,et al.Statistical confidence for likelihood-based paternity inference in natural populations[J].Mol Ecol,1998,7(5):639-655.DOI: 10.1046/j.1365-294x.1998.00374.x.
[16]BASAK S,RAMESH A M,KESARI V,et al.Genetic diversity and relationship of Hedychium from northeast India as dissected using PCA analysis and hierarchical clustering[J].Meta Gene,2014,2:459-468.DOI: 10.1016/j.mgene.2014.05.002.
[17]LIU K J,MUSE S V.PowerMarker:an integrated analysis environment for genetic marker analysis[J].Bioinformatics,2005,21(9):2128-2129.DOI: 10.1093/bioinformatics/bti282.
[18]PRITCHARD J K,STEPHENS M,DONNELLY P.Inference of population structure using multilocus genotype data[J].Genetics,2000,155(2):945-959.DOI: 10.1093/genetics/155.2.945.
[19]李清,罗永坚,吴柔贤,等.广东省大豆种质资源遗传多样性分析及DNA分子身份证构建[J].广东农业科学,2020,47(12):221-228.LI Q,LUO Y J,WU R X,et al.Analysis on genetic diversity and construction of DNA molecular identity card of soybean germplasm resources in Guangdong Province[J].Guangdong Agric Sci,2020,47(12):221-228.DOI: 10.16768/j.issn.1004-874x.2020.12.023.
[20]BOTSTEIN D,WHITE R L,SKOLNICK M,et al.Construction of a genetic linkage map in man using restriction fragment length polymorphisms[J].Am J Hum Genet,1980,32(3):314-331.
[21]EVANNO G,REGNAUT S,GOUDET J.Detecting the number of clusters of individuals using the software STRUCTURE:a simulation study[J].Mol Ecol,2005,14(8):2611-2620.DOI: 10.1111/j.1365-294X.2005.02553.x.
[22]陶乃奇,张斌,刘信凯,等.利用荧光标记SSR鉴别21个茶花新品种[J].植物学报,2019,54(1):37-45.TAO N Q,ZHANG B,LIU X K,et al.Identification of 21 new Camellia hybrid varieties by fluorescence-labelled simple sequence repeat markers[J].Chin Bull Bot,2019,54(1):37-45.DOI: 10.11983/CBB18019.
[23]ZHAO Y N,WANG Y,WANG L X,et al.Molecular identification of mung bean accessions (Vigna radiata L.) from Northeast China using capillary electrophoresis with fluorescence-labeled SSR markers[J].Food Energy Secur,2020,9(1):e182.DOI: 10.1002/fes3.182.
[24]黄兴发,尹跃,赵建华,等.黑果枸杞基因组SSR标记开发及遗传多样性分析[J].西北农林科技大学学报(自然科学版),202 49(1):126-135.HUANG X F,YIN Y,ZHAO J H,et al.Development of genomic SSR markers and genetic diversity analysis of Lycium ruthenicum Murr[J].J Northwest A F Univ (Nat Sci Ed),202 49(1):126-135.DOI: 10.13207/j.cnki.jnwafu.2021.01.015.
[25]AVVARU A K,SHARMA D,VERMA A,et al.MSDB:a comprehensive,annotated database of microsatellites[J].Nucleic Acids Res,2020,48(D1):155-159.DOI: 10.1093/nar/gkz886.
[26]杨凯敏,李贵全,郭数进,等.大豆自然群体SSR标记遗传多样性及其与农艺性状的关联分析[J].核农学报,2014,28(9):1576-1584.YANG K M,LI G Q,GUO S J,et al.Genetic diversity and association analysis of agronomic traits with SSR in a natural population of soybean cultivars[J].J Nucl Agric Sci,2014,28(9):1576-1584.DOI: 10.11869/j.issn.100-8551.2014.09.1576.
[27]OHTSUBO K,NAKAMURA S.Cultivar identification of rice (Oryza sativa L.) by polymerase chain reaction method and its application to processed rice products[J].J Agric Food Chem,2007,55(4):1501-1509.DOI: 10.1021/jf062737z.
[28]DANGL G S,YANG J,GOLINO D A,et al.A practical method for almond cultivar identification and parental analysis using simple sequence repeat markers[J].Euphytica,2009,168(1):41-48.DOI: 10.1007/s10681-008-9877-0.
[29]陈昌文,曹珂,王力荣,等.中国桃主要品种资源及其野生近缘种的分子身份证构建[J].中国农业科学,201 44(10):2081-2093.CHEN C W,CAO K,WANG L R,et al.Molecular ID establishment of main China peach varieties and peach related species[J].Sci Agric Sin,201 44(10):2081-2093.DOI: 10.3846/j.issn.0578-175.2011.10.013.
(责任编辑 吴祝华)
