从肠促胰岛素发现到神奇减肥药诞生
2024-12-25郭晓强
新陈代谢是生命的基本特征,是细胞进行能量生成和利用、结构物质更新等过程的基础。糖是能量生成的主要物质来源,足量的糖对器官发育和机体健康至关重要。低血糖可导致能量供应不足而危害器官的功能,对大脑尤其明显,低血糖易引发头晕、嗜睡、幻觉甚至休克。长期的高血糖(如糖尿病)会造成血管、神经、眼和肾等器官组织不可逆的损伤,进而出现诸多并发症,甚至危及生命。此外,糖摄入过多还是造成肥胖的重要原因。因此稳定的血糖水平具有十分重要的意义,该过程主要由内分泌系统调节。
内分泌系统和血糖调节
1902年,英国生理学家贝利斯(W. M. Bayliss)和斯塔林(E. H. Starling)将肠壁提取物注射到狗体内引起胰液分泌增加,这说明肠道可产生具有调节功能的物质,他们将其命名肠道分泌素(secretion)。现已知盐酸从胃进入十二指肠后刺激肠道分泌素合成并释放入血,最终促进胰腺导管分泌水和碳酸氢盐,使盐酸迅速稀释、中和,避免损害肠道内壁。
1906年,斯塔林提出“激素(hormone)” (又译作荷尔蒙)一词,指代一类身体特定部位(内分泌腺)在外界影响下释放到血液的、含量稀少却可高效影响远距离靶器官生物学作用的物质。激素的发现标志着内分泌学的诞生,在推动学科发展同时,也为临床应用奠定了基础。
19世纪下半叶,有人观察到切除胰腺可造成狗出现糖尿病症状,提示胰腺会产生降血糖物质,该物质被命名为胰岛素。然而,胰岛素的分离却迟迟未获成功,因为胰腺兼具外分泌(分泌蛋白酶入肠道)和内分泌(分泌胰岛素入血)功能,而蛋白酶会破坏胰岛素,使分离失败。1921年,加拿大生理学家班廷(F. G. Banting)等采用胰腺结扎破坏外分泌腺功能,成功分离得到胰岛素,在糖尿病动物和患者体内证明了其降血糖的作用,胰岛素成为治疗糖尿病的基本药物。班廷因此分享了1923年诺贝尔生理学或医学奖。
与此同时,各国科学家也积极开展胰岛素研究。1922年底,美国生理学家金博尔(C. P. Kimball)和默林(J. R. Murlin)采用班廷的策略,在得到胰岛素的同时还意外发现一种升高血糖成分,称胰高血糖素(glucagon)[1]。因此,胰腺是维持血糖恒定的主要内分泌器官,其A细胞和B细胞分别合成胰高血糖素和胰岛素,它们通过相互拮抗方式实现其生理效应。但后续研究发现肠道也拥有血糖调节功能。
肠道内分泌
很早就发现一种现象,口服葡萄糖提升血糖的作用明显弱于静脉注射葡萄糖,但背后机制一直不详。1932年,比利时生理学家拉·巴雷(J. La Barre)提出肠道内分泌假说,认为口服葡萄糖会刺激肠道特定细胞合成肠促胰岛素(incretin),该激素释放入血后作用于胰腺B细胞,增加胰岛素分泌,实现降血糖目的,故口服葡萄糖对提升血糖的效果不及葡萄糖静脉输入。尽管肠道提取物注射到动物血液中具有一定的降血糖效果,但由于血液中的胰岛素含量极低,常规方法难以检测到,无法判定是否为肠促胰岛素的作用。
1950年代底,放射免疫测定(radioimmunoassay, RIA)的发明解决了对胰岛素和胰高血糖素等精确测定的难题。1964—1967年,3个研究小组独立发现,口服葡萄糖比静脉输入相同量的葡萄糖造成的血液胰岛素浓度升高更明显,从而证实了肠促胰岛素效应的存在。
1971年,英国内分泌学家布朗(J. C. Brown)从肠黏膜分离到一种激素,注入动物体内可抑制胃酸分泌,故称胃抑制多肽(gastric inhibitory polypeptide, GIP)。进一步发现它还可增加血液胰岛素含量,后改名葡萄糖依赖型促胰岛素肽(glucose-dependent insulinotropic peptide, GIP)。
后续研究发现,GIP主要在小肠近端十二指肠和空肠内分泌细胞(K细胞)中合成,禁食状态下分泌较少,当口服含葡萄糖或脂肪食物后释放到血液中含量迅速增加,通过刺激胰岛素生成而发挥降血糖作用。这些结果表明,GIP符合肠促胰岛素特性,但遗憾的是,随后用于2型糖尿病治疗时并未获理想效果,最终失败。
大量实验显示,GIP并不是唯一的肠促胰岛素,因为去除GIP后的肠道提取物,仍有促胰岛素的作用,从而促使研究人员继续寻找。
胰高血糖素样肽
1970年代,年轻的丹麦住院医生霍尔斯特(J. J. Holst)发现,接受胃切除术的患者在餐后出现血液胰岛素飙升和血糖急剧下降的现象,推测是因为肠促胰岛素的缘故。当时已知胰高血糖素具有促进胰岛素合成的效果,因此霍尔斯特推测肠道可能也合成胰高血糖素。早在1966年,研究人员就发现肠道存在胰高血糖素阳性信号,霍尔斯特进一步利用胰高血糖素抗体,采用免疫组化方法在肠道L细胞上证实了这一结果,但深入研究发现,那并非胰高血糖素,而是肠高血糖素(glicentin),它由69个氨基酸构成,其中33—61位氨基酸正好对应胰高血糖素的29个氨基酸。胰高血糖素基因首先翻译出胰高血糖素前体蛋白,在胰腺A细胞经过加工产生胰高血糖素,在小肠L细胞经过加工产生肠高血糖素,肠高血糖素还可进一步加工产生胃泌酸调节素(oxyntomodulin)。深入分析发现,肠高血糖素并不促进胰岛素分泌,胃泌酸调节素具有一定促进活性,被列为候选者。由于肠高血糖素和胰高血糖素都含胃泌酸调节素序列,因此RIA不可避免产生交叉反应,最终借助液相和质谱方法将其排除。肠促胰岛素的寻找再次陷入困境。
1980年代初,DNA重组和测序技术的发明和应用推动了内分泌学的快速发展。1981年,美国麻省总医院分子内分泌实验室主任哈贝纳(J. F. Habener)团队获得琵琶鱼(anglerfish)胰高血糖素前体蛋白基因,推导出编码的蛋白质氨基酸序列,发现其N端对应肠高血糖素,但C端还拥有一个额外片段,其序列与胰高血糖素具有较高同源性,称之为胰高血糖素样肽(glucagon-like peptide, GLP)。不久,又获得人、仓鼠、牛和大鼠等哺乳动物的胰高血糖素基因,发现有两种GLP,GLP-1和GLP-2。两种GLP的发现为肠促胰岛素研究带来新机会。
霍尔斯特也发现猪和人的胰高血糖素前体蛋白可在肠道中加工出GLP-1和GLP-2并分泌入血液,但该过程并不存在胰腺。最初推测GLP-1对应胰高血糖素前体蛋白72-108位氨基酸(37个氨基酸),GLP-2对应126-158位(33个氨基酸)。再次令人失望的是,使用这两种天然形式的GLP均未表现出促胰岛素生成的效应,但于1986年底从猪小肠黏膜中分离出一种N端缺少6个氨基酸的GLP-1(7-37)[2],进一步的实验证明其具有强烈的促胰岛素生成效应。几乎在同时,哈贝纳小组也得到同样的结论。

1984年,年轻的加拿大内分泌学家德鲁克(D. J. Drucker)加入哈贝纳实验室,被分派负责GLP研究,同时多肽研究专家莫伊索夫(S. Mojsov)加入项目组。莫伊索夫首先用化学方法合成不同长度GLP-1,包括GLP-1(1-37)和GLP-1(7-37),进一步制备出针对不同GLP-1的抗体,随后采用免疫组化方法检测它们在体内的分布,结果发现大鼠肠道存在的是GLP-1(7-37),基于这一事实推测它可能是GLP-1的活性形式。德鲁克使用大鼠胰岛细胞在体外证明GLP-1(7-37)可促进胰岛素的合成和分泌[3]。莫伊索夫用大鼠模型确定注射GLP-1(7-37)可显著增加血液的胰岛素含量,并且二者的浓度呈正相关[4]。1987年底英国的布鲁姆(S. R. Bloom)团队在人体内也证实了这一结论。至此,GLP-1(7-37)[因最后一位氨基酸通常为酰胺形式,有时也写作GLP-1(7-36)]确定是一种重要的肠促胰岛素,为简便起见,通常直接写作GLP-1。
GLP-1药物研发
产生GLP-1的L细胞主要位于小肠远端,甚至还包括结肠,可诱导GLP-1合成并释放入血的物质主要为单糖(包括葡萄糖、半乳糖和果糖等),此外还有氨基酸和脂肪酸等。GLP-1发挥作用依赖于靶细胞的膜上受体,其受体分布于胰腺细胞、胃肠道、中枢神经系统(包括下丘脑、孤束核和纹状体等)、心脏、肺和肾等多个组织或器官,提示GLP-1具有生物功能多样性[5]。GLP-1除促进胰岛素合成和分泌外,还具有抑制胰高血糖素分泌、降低胃排空和动力、产生饱腹感、降低血压、炎症和凝血、促进排钠和排尿等作用,这预示着它具有宽广的临床治疗价值。
GLP-1前景最初并不被看好,主要源于GIP失败的教训,但其独特的性质仍促使研究人员进行尝试。1992年,将人工合成GLP-1给予1型和2型糖尿病患者,可在不需胰岛素情况下实现血糖正常化,另一项研究表明GLP-1可有效降低2型糖尿病患者餐后血糖水平;第二年,进一步的研究表明静脉注射GLP-1可使运用其他方法控制不佳的2型糖尿病患者空腹和餐后血糖正常化,同时不引起低血糖。这些结果提振了GLP-1相关药物的研发信心,诺和诺德、阿斯利康和礼来等公司纷纷聚焦于此。
但在实际研发过程中却面临众多难题。GLP-1和胰岛素类似,也是一种多肽,通常为皮下注射,但其在体内的半衰期极短,通常只有2分钟,为此研究人员采用连续6周皮下注射GLP-1方式进行测试,结果使严重2型糖尿病患者空腹血糖和餐后血糖明显降低,糖化血红蛋白数据也得到明显改善,最重要的是无明显不良反应。深入的研究揭示了天然GLP-1半衰期短的两个主要原因。首先是体内酶的作用,GLP-1容易被血液中的二肽基肽酶-4(dipeptidyl peptidase-4, DPP-4)识别并降解,导致活性丧失;其次是被肾脏滤过排出体外,解决这两个问题成为随后药物研发的起点,而基于此原理开发的药物称为GLP-1受体激动剂(GLP-1 receptor agonist, GLP-1 RA)[6]。
研究人员从一种毒蜥蜴唾液中分离到毒蜥外泌肽-4(exendin-4),它与人GLP-1具有53%的同源性,在体内可高效激活胰岛素分泌。毒蜥外泌肽-4对DPP-4不敏感,半衰期30分钟,在体内发挥活性时间可5~7小时,这一特性引起了制药公司的注意。阿斯利康公司最终将毒蜥外泌肽-4开发为药物,称艾塞那肽(exenatide)。Ⅲ期临床试验显示,艾塞那肽应用30周后会显著改善空腹血糖和糖化血红蛋白的指标。

诺和诺德公司克努森(L. B. Knudsen)领导的团队则采取另一种策略,在人GLP-1的序列稍加修饰的基础上,与棕榈酰连接研发成功利拉鲁肽(liraglutide)。利拉鲁肽既免遭DDP-4降解,又减少肾脏清除,使半衰期延长到12小时,体内活性持续24小时。Ⅲ期临床试验显示经过为期5周的利拉鲁肽治疗,可更加显著降低空腹血糖水平,并意外发现可使体重减轻2.4%。
礼来公司也成功开发出GLP-1受体激动剂杜拉鲁肽(dulaglutide)。杜拉鲁肽在采用GLP-1的3个氨基酸替换(避免DDP-4降解、增加溶解性和减少免疫原性)基础上,外加连接免疫球蛋白G4(immunoglobulin G4, IgG4)的fc片段,最大限度地保留了药物活性,同时提升稳定性。Ⅲ期临床试验也证实有效性。此外,多家公司也开发成功自己的GLP-1受体激动剂,增加了治疗选择[7]。
GLP-1药物的临床应用
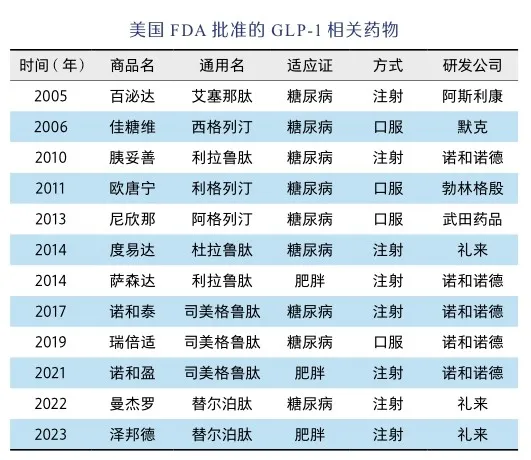
美国FDA先后批准多款GLP-1受体激动剂药物应用于2型糖尿病治疗,包括2004年推出的第一款艾塞那肽(商品名百泌达),2010年的利拉鲁肽(商品名胰妥善),2014年的杜拉鲁肽(商品名度易达)等,这些药物均为皮下注射,外加饮食控制和运动。诺和诺德在利拉鲁肽的基础上改进研发成功司美格鲁肽(Semaglutide),进一步增加了其稳定性,实现每周皮下注射1次,于2017年被FDA批准用于2型糖尿病治疗(商品名诺和泰)。对于长期用药的患者而言,皮下注射会带来极大不便,诺和诺德又开发成功口服型司美格鲁肽,于2019年批准用于2型糖尿病治疗(商品名瑞倍适),每天一次。
礼来公司开发成功靶向GLP-1和胰高血糖素受体的双重激动剂替尔泊肽(Tirzepatide),于2023年被FDA批准应用于2型糖尿病治疗(商品名曼杰罗)。GLP-1受体激动剂在应用于糖尿病治疗的过程中,还被发现有其他药效,其中最重要的是肥胖。
随着经济发展和生活水平提高,肥胖发生率逐年增加,相对于多药可用的糖尿病,肥胖一直缺乏安全有效的药物,GLP-1受体激动剂的应用改变了这一状况[8]。GLP-1受体激动剂可跨越血脑屏障进入大脑而影响饮食,在保障饥饿性饮食(维持个体生命)基础上最大程度减少奖励性饮食(获得愉悦感),相当于借助药物达到抵制美食诱惑的目的。2014年,利拉鲁肽成为第一款被FDA批准的GLP-1减肥药(商品名萨森达),每天注射1次。2021年,司美格鲁肽被批准用于肥胖/超重人群使用(商品名诺和盈),每周注射1次。2023年,礼来公司替尔泊肽也被批准用于减肥(商品名泽邦德),每周注射1次。这些药物的应用和改进,在增强患者舒适度基础上还极大提升了肥胖治疗效果。多款GLP-1受体激动剂的批准应用成功解决多年来肥胖治疗无药可用的窘迫状况。
除以上这些GLP-1受体激动剂外,DDP-4抑制剂是另一个药物发展方向,它们都是小分子物质,采用口服方式。默克公司开发成功西格列汀(Sitagliptin)(商品名佳糖维),于2006年被FDA批准用于2型糖尿病辅助治疗(通常联合二甲双胍等药物),每日1次。随后,多款DDP-4抑制剂被研发成功应用于临床,包括沙格列汀(Saxagliptin)、利格列汀(Linagliptin)和阿格列汀(Alogliptin)等[9]。
迄今已有超过1亿2型糖尿病患者接受GLP-1受体激动剂或DPP-4抑制剂治疗, GLP-1药物销售额仅2021年就达220亿美元。GLP-1药物在肥胖治疗中的应用更是成为近两年风靡全球的话题,掀起了减肥新热潮,不仅改观肥胖治疗方式,而且革新了对肥胖的认识——肥胖是一种慢性病,除锻炼和饮食控制外,还需辅助药物治疗。
目前尚有多款药物还在临床试验阶段,有望进一步提升疗效。更重要的是,这些药物的适应证也在逐渐拓展,目前发现对心血管疾病(心肌梗死和动脉粥样硬化)、神经退行性疾病(阿尔茨海默病和帕金森病)、糖尿病肾病、代谢性肝病和炎症等也拥有潜在治疗价值[10]。
GLP-1的重大价值
GLP-1的发现及其作用机制的阐明是近几十年来内分泌学乃至医学领域的最重大进展之一,基于GLP-1的治疗方法更是在提升2型糖尿病和肥胖等疗效方面发挥了重要作用,GLP-1相关药物被《科学》周刊评为2023年度十大科学突破之首。
随着GLP-1重要性的体现,做出奠基性贡献的几位科学家获得了许多重大科学奖项,如霍尔斯特、哈贝纳和德鲁克等,先后分享沃伦·阿尔珀特基金会奖(2020年)、盖尔德纳国际奖(2021年)、阿斯图里亚斯亲王科学技术奖(2024年)等,霍尔斯特、哈贝纳和莫伊索夫分享唐奖(2024年),哈贝纳、莫伊索夫和克努森分享拉斯克-德贝基临床医学研究奖(2024年)。GLP-1生理作用和药用价值的重要性,无疑也使几位科学家成为近几年诺贝尔生理学或医学奖的热门候选人。
[1]Kimball C P, Murlin J R. Aqueous Extracts of Pancreas Ⅲ. Some Precipitation Reactions of Insulin. J Biol Chem, 1923, 58(1):337–346.
[2]Holst J J, Orskov C, Nielsen O V, et al. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 1987, 211(2): 169-174.
[3]Drucker D J, Philippe J, Mojsov S, et al. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci USA, 1987, 84 (10): 3434–3438.
[4]Mojsov S. Weir G C, Habener J F. Insulinotropin: glucagonlike peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest, 1987, 79 (2): 616–619.
[5]Holst J J. The physiology of glucagon-like peptide 1. Physiol Rev, 2007, 87(4): 1409-1439.
[6]Drucker D J, Habener J F, Holst J J. Discovery, characterization, and clinical development of the glucagon-like peptides. J Clin Invest, 2017, 127(12): 4217-4227.
[7]Drucker D J. The GLP-1 journey: From discovery science to therapeutic impact. J Clin Invest, 2024, 134(2): e175634.
[8]Jastreboff A M, Kushner R F. New frontiers in obesity treatment: GLP-1 and nascent nutrient-stimulated hormone-based therapeutics. Annu Rev Med, 2023, 74: 125-139.
[9]O’Rahilly S. The islet’s bridesmaid becomes the bride: Proglucagon-derived peptides deliver transformative therapies. Cell, 2021, 184(8): 1945-1948.
[10]Drucker D J. The benefits of GLP-1 drugs beyond obesity. Science, 2024, 385(6706): 258-260.
关键词:肠道内分泌 胰高血糖素样肽-1 代谢性疾病 受体激动剂 ■
