区分肺腺癌和肺鳞状细胞癌的潜在基因分析
2024-05-11朱德奇卢建国李宾张劲草王忠民董成来
朱德奇,卢建国,李宾,张劲草,王忠民,董成来
(新乡医学院第一附属医院 胸外科一病区,河南 新乡 453100)
肺癌是世界上发病率和病死率最高的恶性肿瘤之一[1]。非小细胞肺癌(non-small cell lung cancer,NSCLC)是肺癌的主要病理类型[2],其病程隐匿,多数患者就诊时病变已进展至中晚期,5 a生存率仅为15.9%[3]。肺腺癌(lung adenocarcinoma,LUAD)和肺鳞状细胞癌(lung squamous cell carcinoma,LUSC)是NSCLC的两种主要组织学亚型[4]。治疗前明确病理分型对中晚期肺癌的治疗至关重要。但除了病理检查外,LUAD和LUSC仍难以区分。基因突变是NSCLC发生发展的重要原因,一些关键基因如NF1、CDKN2A和TP53在LUAD和LUSC中均具有较高的突变率[5]。然而LUAD和LUSC中也存在着不同的驱动基因[6],发现独特的可变基因有助于区分NSCLC亚型,及时提供合适的治疗方案。生信分析广泛应用于发掘肿瘤差异表达的靶向治疗基因[7]。鉴于目前缺乏有效区分LUAD和LUSC的潜在基因,本研究采用生信手段,通过分析基因表达综合(Gene Expression Omnibus,GEO)和肿瘤基因组图谱(The Cancer Genome Atlas,TCGA)数据库中的NSCLC数据集,旨在寻找能有效区分LUAD和LUSC的潜在基因。
1 材料和方法
1.1 GEO数据集
美国国家生物技术信息中心的GEO数据库(http://www.ncbi.nlm.nih.gov/geo/)是一个提供基因表达、芯片和微阵列数据的基因组公共数据存储库。纳入研究的3个数据集GSE4882、GSE7339和GSE40275同时包含LUAD和LUSC的RNA-seq数据。其中GSE4882包括60个LUAD和52个LUSC组织样本;GSE7339包括32个LUAD和12个LUSC组织样本;GSE40275包括11个LUAD和5个LUSC组织样本。
1.2 筛选差异表达基因(differentially expressed genes,DEGs)
利用R软件对GEO数据库下载的mRNA原始微阵列基因表达数据进行处理。使用Limma包对数据进行归一化处理并进行差异分析,P<0.05且|log2FC|>1的基因为DEGs,利用R软件绘制韦恩图,3个GEO数据库交叉得到共同的DEGs。
1.3 基因本体(Gene Ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)分析
GO富集分析包括生物通路、分子功能和细胞成分分析3个方面内容。KEGG通路富集分析用于DEGs所富集的通路注释分析。GO富集分析和KEGG分析用于探究DEGs的潜在功能。GO富集分析和KEGG分析由DAVID数据库完成。P<0.05为差异有统计学意义。
1.4 构建蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络,识别枢纽基因
利用STRING在线数据库(http://string-db.org/)构建PPI网络,利用Cytoscape软件重建PPI网络,找出DEGs编码蛋白之间的相关性,分析得到枢纽基因。
1.5 TCGA数据库
TCGA是包括几乎所有肿瘤RNA-seq数据的数据库之一。利用在线网站GEPIA (http://www.gepia.cancer-pku.cn/)分析TCGA数据库中枢纽基因的表达。采用ULACAN在线工具分析TCGA数据库中枢纽基因表达与预后风险的关系。P<0.05为差异有统计学意义。
1.6 构建风险预测模型
利用R软件进行Cox回归分析和Nomogram列线图的构建。Cox回归分析用于计算枢纽基因与患者死亡的风险比。建立Cox比例风险模型Nomogram来预测枢纽基因的风险值。使用校准曲线对构建的模型进行验证。一致性指数用于评价预后模型的预测精度,0.50~0.70为准确度较低,0.71~0.90为准确度中等,高于0.90为准确度高。
1.7 组织标本
本研究收集的12例新鲜肺癌组织标本均为2023年4—6月新乡医学院第一附属医院胸外科手术切除的患者组织。术后病理诊断为LUSC或LUAD,术前均未接受针对肿瘤的任何治疗。组织标本离体后置于40 g·L-1的多聚甲醛中保存。
1.8 RNA提取和qRT-PCR
使用TRIzol试剂提取组织总RNA,提取过程全程无酶操作。利用逆转录试剂(Takara,Japan)将mRNA逆转录为cDNA,随后使用qRT-PCR试剂盒(Takara,Japan)将上述反转录过程中得到的cDNA进行PCR扩增和定量分析,每个样本设置3个重复孔。GAPDH作为正常对照,按2-ΔΔCt法计算RNA表达量。引物序列:KRT18(正义链5’-TGCAGCTGGAGACAGAAATC-3’,反义链5’-TTCCACAGTCAACCCAGAAC-3’);GAPDH(正义链5’-ACAGCAACAGGGTGGTGGAC-3’,反义5’-TTTGAGGGTGCAGCGAACTT-3’)。
1.9 Western-blot
用RIPA裂解缓冲液提取组织蛋白。利用SDS-PAGE凝胶(100 g·L-1)电泳,然后将凝胶转移到NC膜(Darmstadt,Germany)上进行转膜。使用无蛋白快速阻断缓冲液封闭NC膜。然后将膜与KRT18(1∶1 000)和GAPDH(1∶1 000)的一抗在4 ℃下孵育过夜。第2天用1×TBST洗涤膜3次。然后,将膜与匹配的二抗(Beyotime,China)在常温下孵育1.5 h。最后显影,记录数据。
1.10 统计学分析
采用SPSS 19.0软件进行统计学分析。采用χ2检验或Fisher确切概率法分析枢纽基因表达与临床病理特征之间的关系;Student’st检验用于比较两组独立的数据;ANOVA检验用于比较多组数据;采用单因素和多因素Cox回归模型进行单因素和多因素分析。P<0.05为差异有统计学意义。
2 结果
2.1 鉴定DEGs
下载分析GSE4882、GSE7339和GSE40275的RNA-seq数据。结果显示在3个数据集中有9个DEGs表达上调(图1A),13个DEGs表达下调(图1B)。其中上调的DEGs是在LUAD中表达上调,在LUSC中下调。下调的DEGs是在LUAD中表达下调,在LUSC中表达上调。
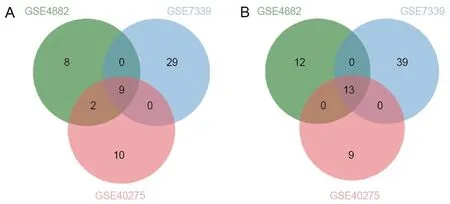
A为韦恩图显示表达上调的DEGs;B为韦恩图显示表达下调的DEGs。图1 韦恩图显示表达上调的DEGs
2.2 GO和KEGG富集分析
利用DAVID数据库对DEGs进行GO和KEGG分析。DEGs可能相关的细胞功能和通路见图2。
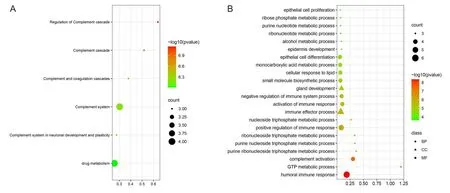
A为散点图显示DEGs的GO富集;B为散点图显示DEGs的KEGG富集。图2 DEGs可能相关的细胞功能和通路
2.3 PPI网络构建
使用在线网站STRING进行PPI蛋白互作分析,分析找到DEGs之间相关联的枢纽基因。然后利用Cytoscape软件重建PPI网络,进一步显示关键枢纽基因(图3)。最后在22个DEGs中发现了KRT18、RAN、NME1、NME2、MIF和CFB6个关键枢纽基因。
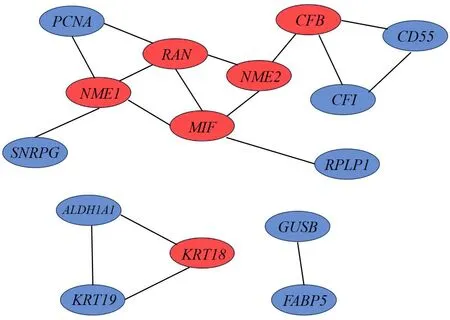
图3 构建DEGs的PPI网络基因分析
2.4 枢纽基因在TCGA数据库中的表达
进一步利用TCGA公共数据库分析上述枢纽基因在LUSC与LUAD患者癌组织样本与癌旁正常组织中的表达差异情况,结果显示,KRT18在LUAD组织中的表达高于癌旁正常组织,在LUSC中则无表达差异;而RAN表达则恰恰相反,其在LUSC组织中的表达高于癌旁组织,但在LUAD中则无表达差异。NME1、NME2、MIF在LUSC及LUAD中表达均高于癌旁组织,而CEB在两种组织中的表达无明显差异(图4)。提示KRT18和RAN可能是LUAD和LUSC分类的潜在基因。
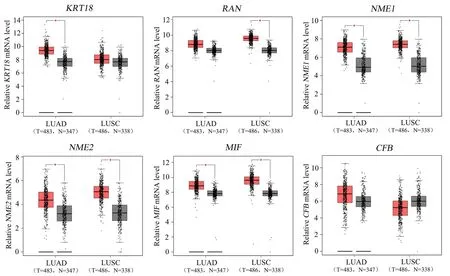
图4 利用GEPIA在线网站分析6个枢纽基因在TCGA数据库中的表达情况
2.5 单因素和多因素Cox分析
利用ULACAN在线工具分别下载TCGA中LUAD和LUSC上述6个枢纽基因的表达量、临床病理特征以及生存数据进行单因素和多因素Cox分析,评估枢纽基因表达与患者生存时间之间的关系。由于CFB在LUAD和LUSC中的表达情况无差异,因此未将CFB纳入单因素和多因素Cox回归分析中。在LUAD中,单因素Cox分析结果显示,KRT18、RAN、NME1、NME2和MIF均是导致LUAD患者死亡的危险因素,进一步多因素Cox回归分析显示,KRT18高表达可作为判断LUAD患者预后的独立危险因素(表1)。而在LUSC患者中,单因素和多因素Cox回归分析显示,KRT18、RAN、NME1、NME2和MIF基因与LUSC患者死亡之间并无显著相关性(表2)。提示KRT18不仅在LUAD和LUSC患者中表达存在差异,在预测死亡上也存在显著差异。因此,推测枢纽基因KRT18可能是区分LUAD和LUSC的潜在基因。
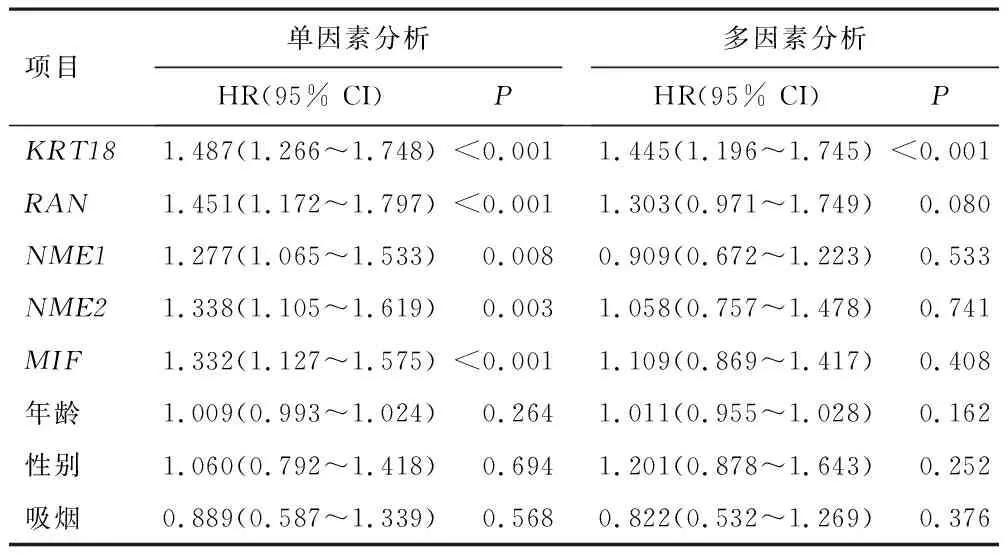
表1 LUAD中枢纽基因表达与临床病理特征以及生存关系的单因素和多因素Cox分析
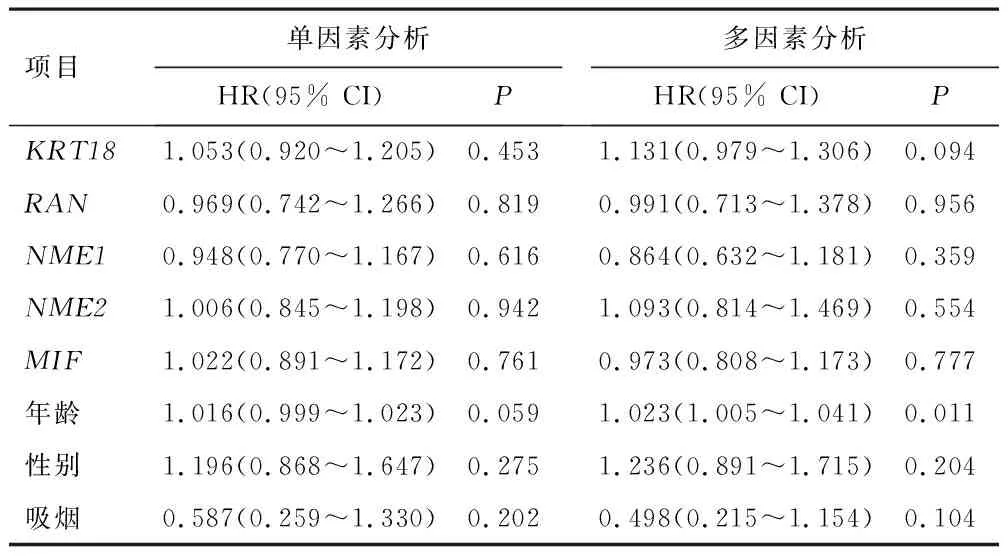
表2 LUSC中枢纽基因表达与临床病理特征以及生存关系的单因素和多因素Cox分析
2.6 构建风险回归模型
本研究构建了KRT18表达对LUAD患者1、2和3 a总生存期影响的Nomogram列线图,对Cox回归分析结果进行可视化呈现(图5A)。此外,建立校正曲线对列线图进行进一步的验证(图5B)。

A为列线图;B为校准曲线。图5 Nomogram列线图和校准曲线的构建
2.7 KRT18表达验证
收集新乡医学院第一附属医院6对LUAD和LUSC患者的肿瘤组织和癌旁组织,分别检测这些配对样本中KRT18mRNA和KRT18蛋白的表达水平,结果显示LUAD患者肿瘤组织的KRT18mRNA和KRT18蛋白表达均高于癌旁组织,但在LUSC中,只有部分组织的KRT18mRNA和KRT18蛋白表达高于癌旁正常组织(图6)。提示KRT18可作为区分LUAD和LUSC的潜在基因。
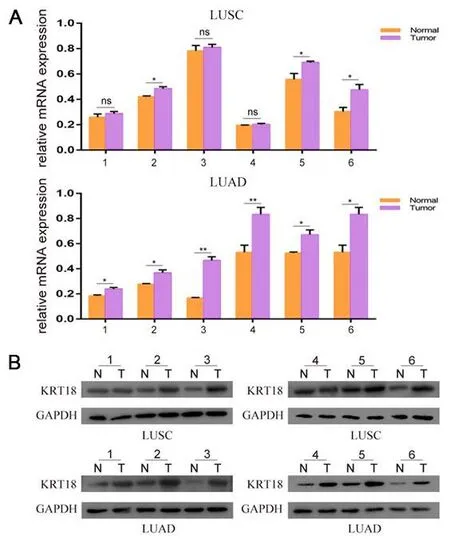
A为KRT18 mRNA表达比较;B为KRT18蛋白表达比较。图6 KRT18在NSCLC临床组织样本中的表达
3 讨论
NSCLC是肺癌的主要病理亚型,约占所有类型肺癌的85%,病程进展隐匿,多数患者就诊时病变已进展至局部晚期,5 a生存率低于20%[3]。研究表明早期诊断及治疗可提升NSCLC患者5 a生存率至57%以上[8]。治疗前明确病理分型和分期对NSCLC治疗至关重要。LUAD和LUSC是NSCLC两种主要的组织学亚型,目前病理检查仍然是明确LUAD和LUSC的金标准,但病理检查需要一段时间,可能延误患者治疗。因此,寻找能区分LUAD和LUSC的基因至关重要。
虽然LUAD和LUSC是不同的NSCLC组织学类型,但研究表明在LUAD和LUSC肿瘤发生发展中发生了许多相似的基因突变。例如CDKN2A、KEAP1和NF1在两种肿瘤类型中均发生突变[5]。TP53是一种传统的抗癌基因,在各种肿瘤的发生中起着重要作用,在LUAD和LUSC中同样会发生突变[9]。EGFR基因突变在LUAD患者中更为常见,而在LUSC患者中则较为罕见[10],这表明发现不同的基因突变可能是帮助LUAD和LUSC分类的潜在方法。本研究通过分析公共数据库中的RNA-seq或基因芯片数据,找到有助于区分LUAD和LUSC潜在的基因。首先,从3个GEO数据库下载RNA-seq数据,分析得到LUAD和LUSC的DEGs。然后,通过构建PPI网络寻找到RAN、NME1、NME2、MIF、CFB和KRT186个枢纽基因,并进一步在TCGA数据库中验证枢纽基因表达,分析枢纽基因表达与临床病理、生存之间的关系,发现KRT18在LUAD和LUSC中的表达与临床病理、生存之间存在显著性差异。因此推测KRT18可能是一个潜在的区分LUAD和LUSC的基因。最后,从收集的LUAD和LUSC患者的临床组织样本中检测了KRT18mRNA和KRT18蛋白水平,结果显示KRT18在不同亚型的NSCLC中表达不同。以上研究结果提示KRT18可以作为区分LUAD和LUSC的潜在基因。KRT18是一种细胞角蛋白,主要保护上皮细胞免受机械和非机械压力作用[11]。角蛋白丝可以动态重塑,在各种机械和非机械刺激下进行重组,调节细胞迁移和信号传导等细胞过程[12]。研究表明KRT18与胃癌、结直肠癌和胶质母细胞瘤等恶性肿瘤的发生和侵袭转移相关[13-15]。此外,另有研究报道EGR1可调控KRT18的表达,介导NCSLC的发生和发展[16]。这些结果表明KRT18是一种致癌基因,可介导多种癌症的发生和发展。
4 结论
本研究证明KRT18可以作为一个潜在的基因来帮助区分LUAD和LUSC,但本试验也存在几点不足,第一,临床组织样本量较小,需要进一步扩大样本量;第二,本试验并未进行KRT18在LUAD以及LUSC中的体内外细胞实验,未来需要做进一步研究证实本研究结论。
