新生儿急性心功能不全1例
2024-05-10李淑涓胡黎园张蓉杨琳奚立刘芳曹云周文浩程国强
李淑涓 胡黎园 张蓉 杨琳 奚立 刘芳 曹云 周文浩 程国强
(国家儿童医学中心/复旦大学附属儿科医院1.新生儿科;2.内分泌遗传代谢科;3.心内科,上海 201102)
1 前言
先天性孤立性促肾上腺皮质激素缺乏症(congenital isolated adrenocorticotropic hormone deficiency, CIAD)是一种罕见的常染色体隐性遗传病,具有临床异质性及家族遗传性,由Steinberg等[1]于1954 年首次报道,该病每年发病率约0.9/100 万人[2]。CIAD 的特点是继发性肾上腺皮质功能不全,促肾上腺皮质激素(adrenocorticotropic hormone, ACTH)和皮质醇分泌减少,垂体其他激素分泌正常或短暂性的可逆性升高,无其他结构性垂体缺陷,并排除外源性糖皮质激素应用以及垂体瘤术后所致的ACTH 缺乏[3]。国内关于CIAD病例报道多以成人或儿童为主,新生儿期诊断CIAD的病例报道目前仅有1例[4]。CIAD典型的临床表现为顽固性低血糖、胆汁淤积和惊厥,而以心功能不全为主要表现的病例暂无报道。CIAD 如未早期识别,可导致新生儿死亡,早期诊断并接受激素替代治疗可显著改善预后。本文描述1 例TBX19基因变异所致以急性心功能不全为主要表现的CIAD患儿的临床诊疗经过,以期提高临床医生对该病的认识。
2 病例介绍
现病史:患儿男,1 日龄,因生后6 h 出现低血糖伴呼吸暂停收入我院新生儿重症监护病房。患儿系第5 胎第2 产,胎龄38+6周,顺产,出生体重2 840 g,无胎膜早破,羊水清,脐带、胎盘未见异常。生后Apgar评分1 min 9分,5 min 9分。生后6 h出现呼吸暂停伴四肢抖动,监测经皮血氧饱和度为75%~80%,心率90~100 次/min,末梢血糖0.9 mmol/L,予10%葡萄糖注射液静脉推注并托背刺激,患儿呼吸无改善,予气管插管常频机械辅助通气、静脉补液及抗感染治疗后,复测末梢血糖3.1 mmol/L,为进一步救治,在气管插管机械通气下转入我院新生儿重症监护病房。患儿母亲正规产检,孕38+4周因“羊水少”于外院住院保胎治疗。患儿父母身体健康,非近亲婚配,否认家族遗传性疾病史。
入院体格检查:神志清楚,反应欠佳。全身皮肤红润,未见色素沉着,无水肿。前囟平软,口唇红润,颈软。气管插管常频机械辅助通气下,吸气性凹陷弱阳性,双肺送气音对称。心音有力,律齐,未闻及杂音。腹平软,肝脾不大,肠鸣音弱。四肢肌张力正常,原始反射可引出,四肢末梢暖,毛细血管充盈时间2 s。
辅助检查:新生儿基因Panel检测(生后第21天) 示TBX19基因存在复合杂合突变(NM_005149, exon7: c.917-2A>G 和 NM_005149,exon4:c.608C>T),该基因为CIAD 致病基因[5]。经父母基因验证,父亲携带c.917-2A>G,母亲携带c.608C>T。余实验室检查结果见表1。心脏彩超(生后第3 天) 示动脉导管未闭(patent ductus arteriosus, PDA)(3.5 mm),射血分数>60%,二尖瓣轻度反流,三尖瓣轻中度反流。心脏彩超(生后第8天)示左室收缩功能减低(射血分数55%),PDA(2.6 mm),二尖瓣中度反流,三尖瓣中重度反流。肝胰肾上腺超声、垂体平扫增强磁共振成像检查未见明显异常。
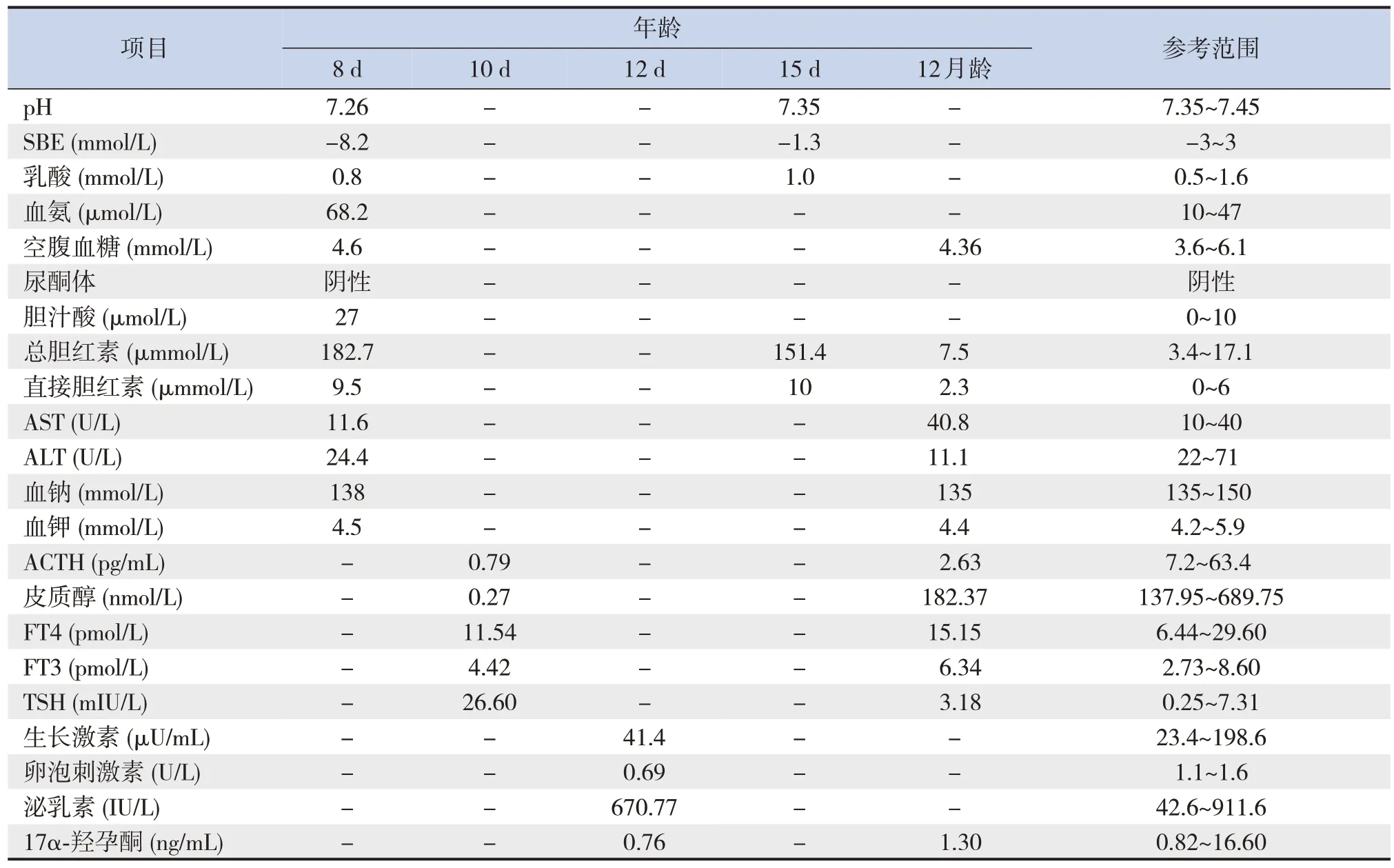
表1 患儿实验室检查结果
3 多学科诊疗
3.1 新生儿重症监护病房初诊
该患儿生后第1天因一过性低血糖及呼吸暂停入院,经治疗后病情趋于稳定。但生后第8天心脏彩超提示左室射血分数降低,同时出现气促及代谢性酸中毒表现,诊断为急性心功能不全。立即予以吸氧、强心治疗,并积极完善相关检查寻找潜在病因。实验室检查示ACTH和皮质醇水平显著降低,考虑患儿存在肾上腺皮质功能不全,推测肾上腺皮质功能不全可能是导致患儿心功能不全的主要原因。
3.2 心内科会诊
多种病因可导致新生儿急性心功能不全,主要可分为结构性心脏病和心肌病2大类(图1)。其中结构性心脏病是新生儿心功能不全的最常见病因。该患儿心脏彩超和心脏增强CT除提示PDA外,未提示其他心脏结构异常。患儿生后早期喂养建立顺利、早期撤离有创机械通气,体格检查示心前区未闻及杂音,不符合血流动力学不稳定PDA,因此不支持由PDA导致的心功能不全。进一步则需考虑是否存在围产期疾病、心律失常、心肌病或全身性疾病引起患儿急性心功能不全。本例患儿皮质醇水平显著降低,皮质醇在维持血管张力和肾上腺素能受体对儿茶酚胺的反应起着非常关键的作用,皮质醇水平不足会导致心率、心肌收缩力下降和血压下降,最终导致心功能不全。本例患儿显著降低的皮质醇水平可能是其急性心功能不全的主要原因。因此治疗上主要为积极治疗原发病,其次可予以强心、利尿、限液等支持治疗。
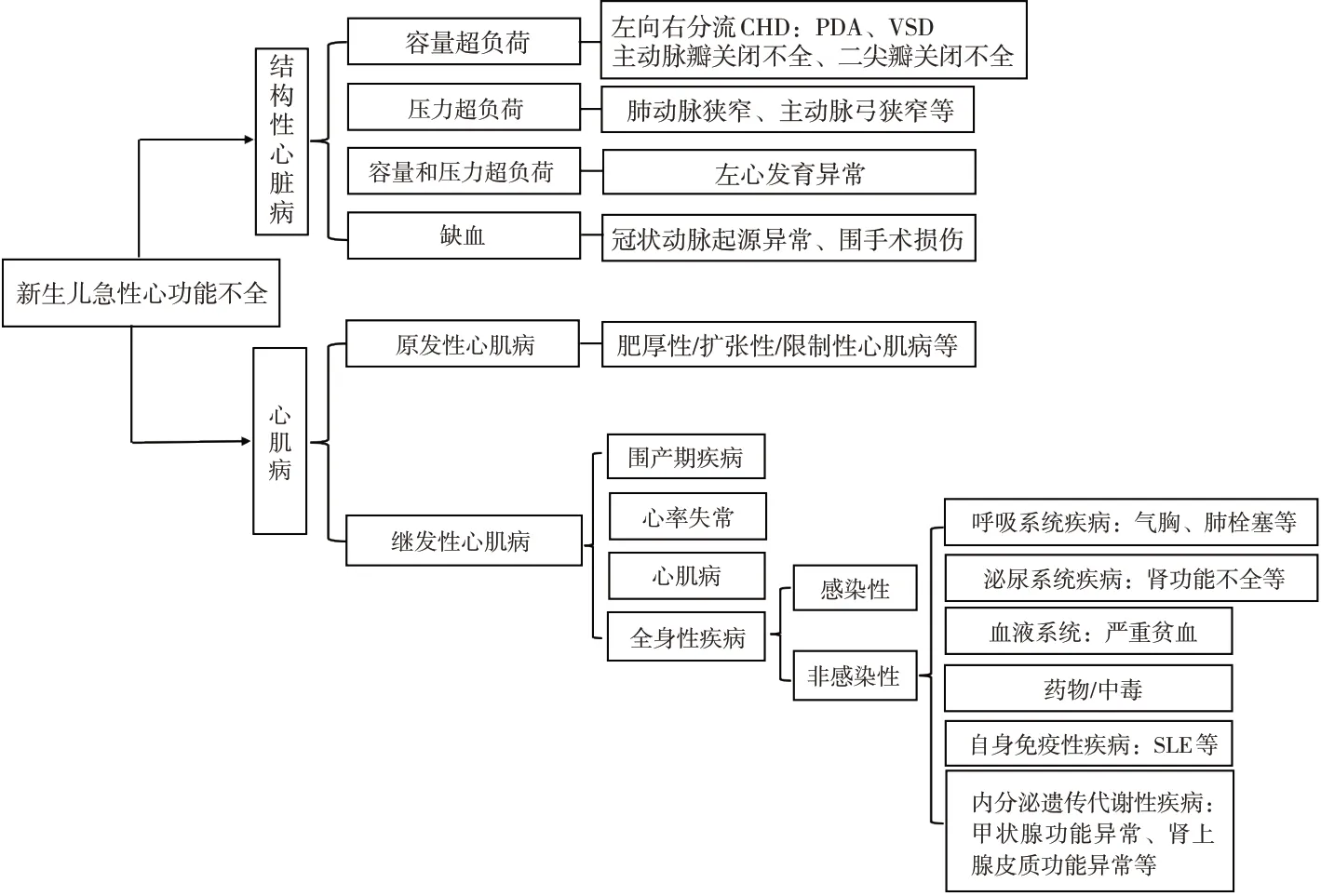
图1 新生儿急性心功能不全的病因鉴别诊断 [CHD]先天性心脏病;[PDA]动脉导管未闭;[VSD]室间隔缺损;[SLE]系统性红斑狼疮。
3.3 内分泌遗传代谢科会诊
患儿以急性心功能不全起病,实验室检查提示ACTH和皮质醇水平显著降低,考虑诊断肾上腺皮质功能不全。患儿出生史及家族史无特殊;生长发育良好,面容无特殊,皮肤无色素沉着;无电解质紊乱、垂体其他激素水平基本正常且垂体磁共振成像无异常,考虑是孤立性ACTH缺乏,新生儿期孤立性ACTH缺乏多是涉及垂体发育的基因突变或基因表达调控异常导致的,建议完善相关基因检测。最终患儿基因检测出TBX19基因存在复合杂合突变,分别来自父母,因此明确诊断TBX19基因变异所致CIAD。CIAD是一种罕见的常染色体隐性遗传病,是由于ACTH缺乏导致继发性肾上腺皮质功能不全,而垂体前叶其他激素水平正常。CIAD 临床分型包括早发型完全性、部分性和晚发型。早发型完全性CIAD 的ACTH 和皮质醇水平更低,均会在新生儿期出现临床表现[6]。CIAD 典型的临床表现为顽固性低血糖、胆汁淤积和惊厥。少数CIAD 病例存在心肌病、Arnold-Chiari Ⅰ型畸形、47,XXX 综合征、单侧后鼻道闭锁[6]。目前以心功能不全为主要表现的新生儿期起病的CIAD,暂未见报道。皮质醇水平可以通过多种机制作用于心血管系统,低水平的皮质醇可能是CIAD导致急性心功能不全的潜在机制。关于新生儿期起病的CIAD治疗文献报道少见。现有文献中CIAD治疗基本均为氢化可的松替代治疗,但氢化可的松替代治疗剂量尚无统一推荐[7-8]。CIAD如未早期识别,可导致新生儿期20%的病死率[9],及时的激素替代治疗有效降低反复低血糖和难治性惊厥的风险,显著改善预后。已有文献报道中大部分新生儿期起病的CIAD经早期氢化可的松替代治疗,预后良好[9-10]。该患儿存在急性心功能不全,病情危重,因此急性期可考虑使用较大剂量[100 mg/(m2·d)]氢化可的松冲击治疗3 d,以尽快控制代谢紊乱,临床症状好转后,尽快减少氢化可的松剂量至维持量[20 mg/(m2·d)]。
3.4 分子诊断中心会诊
TBX19基因变异是早发型完全性CIAD 的最主要原因。垂体分泌的ACTH是在下丘脑促肾上腺皮质激素释放激素作用下,刺激垂体前叶首先合成阿黑皮素原(proopiomelanocortin, POMC),经蛋白质转换酶裂解后生成ACTH。转录TBX19基因对垂体前叶POMC细胞的发育分化至关重要。TBX19基因属于T-box基因家族,TBX19基因可通过T-box基因结构域与POMC启动子结合,激活POMC基因转录,诱导垂体分化表达POMC的细胞系,变异后POMC基因转录减少,影响ACTH产生。该患儿基因检测显示TBX19基因存在复合杂合变异,该变异在HGMD数据库中尚无相关文献报道。通过对患儿父母进行Sanger测序,父亲携带c.917-2A>G,母亲携带c.608C>T,判定为可疑致病变异[11],结合临床表型考虑该2个变异为该患儿的致病原因。
3.5 新生儿重症监护病房诊断思路总结
该患儿急性起病,以心功能不全为主要特征。实验室检查提示ACTH和皮质醇水平显著降低,但垂体磁共振成像正常,基因检测示TBX19基因复合杂合变异,分别来自父母,明确诊断TBX19基因变异所致CIAD,早期予以氢化可的松治疗。以心功能不全为主要表现的CIAD目前未见报道。该患儿临床仅表现为生后早期一过性低血糖及急性心功能不全,住院期间血糖、转氨酶、胆汁酸及直接胆红素水平基本正常,未出现顽固性低血糖、惊厥或胆汁淤积,这可能与早期诊断及早期氢化可的松替代治疗有关。由于该患儿生后早期仅出现过一次低血糖,因此在该病例诊治过程中,极易忽视低血糖的线索。
4 治疗经过及转归
患儿入院后胸部X线片示左侧少量气胸,继续予常频机械辅助通气、静脉补液及抗感染治疗。生后第2天成功撤离呼吸机,病情趋于稳定。生后第8 天复查心脏超声,结果提示左室射血分数55%,PDA(2.6 mm),二尖瓣、三尖瓣中重度反流。之后24 h 内出现气促,血气分析提示代谢性酸中毒。立即予以鼻导管吸氧、纠酸及强心治疗,患儿气促缓解不明显,随访心脏超声提示左室射血分数进行性降低。生后第12 天诊断肾上腺皮质功能不全后予氢化可的松治疗,后临床症状迅速好转。生后第21 天基因诊断结果示TBX19基因的复合杂合突变,突变分别来自父亲和母亲,患儿明确诊断TBX19基因变异所致CIAD。生后第41天患儿顺利出院。患儿住院期间诊疗经过详见图2。出院后继续口服氢化可的松治疗,目前患儿18 月龄,居家监测血糖稳定;无惊厥、胆汁淤积及心功能不全等表现,生长发育良好,12 月龄时门诊随访甲状腺激素及17α-羟孕酮基本正常。
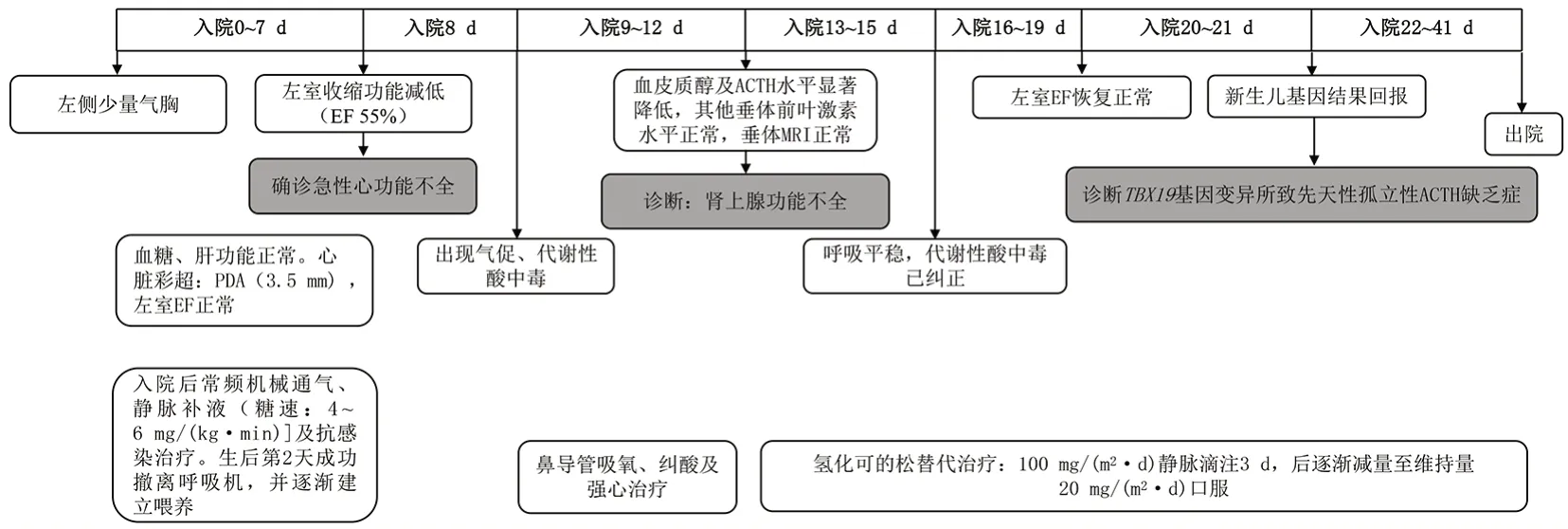
图2 诊疗经过 [EF]射血分数;[ACTH]促肾上腺皮质激素;[MRI]磁共振成像;[PDA]动脉导管未闭。
5 小结
该患儿住院期间突发不明原因急性心功能不全。在病因寻找方面通过多学科诊疗,最后通过激素水平测定、垂体磁共振成像及基因结果,明确诊断TBX19基因突变所致CIAD。早期予以氢化可的松替代治疗,预后良好。以下是我们在本病例诊治过程中的几点体会:(1)新生儿急性心功能不全需警惕内分泌遗传代谢性疾病,应尽早行皮质醇检测。(2)CIAD 如未早期识别,可导致新生儿死亡,针对该疾病,早期诊断及治疗对预后至关重要。(3)CIAD典型临床表现为反复低血糖、胆汁淤积及惊厥,基因检测是确诊依据。部分患儿可无上述典型表现,因此在临床危重症新生儿救治过程中,需警惕该疾病,建议尽早行基因检测。
作者贡献声明:李淑涓负责撰写文章、病例采集与分析;胡黎园、张蓉、杨琳、奚立、刘芳、曹云、周文浩负责病例分析指导;程国强负责文章指导与修改。
利益冲突声明:所有作者声明无利益冲突。
