Ⅰ型神经纤维瘤病合并小肠间质瘤及嗜铬细胞瘤1例报告并文献复习
2024-04-29郑林熠李玉强陶醉李丰源刘伟伍韶斌裴海平
郑林熠,李玉强,陶醉,李丰源,刘伟,伍韶斌,裴海平
[1.中南大学湘雅医院 胃肠外科,湖南 长沙 410000;2.国家老年疾病临床医学研究中心(湘雅医院),湖南 长沙 410008]
Ⅰ型神经纤维瘤病(neurofibromatosis type 1,NF1)又称为von Recklinghausen 病,是一种常染色体显性遗传病,发病率约为1/3 000,发病情况无人种差异[1-2]。NF1 多表现为皮肤多发扁平浅棕色皮肤色素斑块,皮下可见神经纤维瘤和Lisch 结节。NF1 的发病机制是基于NF1 抑癌基因的突变,引起细胞内神经纤维蛋白的产生减少。这导致RAS 基因过度激活,进而导致Akt/mTOR 和Raf/MEK/ERK通路激活ERK,进而激活SYN1,调节GABA 释放[3]。NF1 的临床表现差异较大,包括肿瘤性或非肿瘤性疾病,主要累及不同区域的神经外胚层或间充质来源的组织,如皮肤、骨、眼和中枢神经系统。此外,肿瘤性病变,如神经纤维瘤、恶性周围神经鞘膜瘤(malignant peripheral nerve sheath tumors,MPNSTs)、嗜铬细胞瘤(pheochromocytoma,PHEO)和胃肠 道间质 瘤(gastro-intestinal stromal tumor,GIST),也为NF1 患者常见合并症[4]。然而,合并2 种及2 种以上的肿瘤性病变的病例报告极为罕见。本文报告1 例NF1合并PHEO 及小肠间质瘤(small intestinal gastrointestinal stromal tumor,SIST)患者的诊疗经过并对相关文献进行回顾,现报告如下。
1 病例资料
患者 女,57 岁。2022 年4 月因“体检发现左肾上腺占位”收治入中南大学湘雅医院胃肠外科。患者既往有多发性皮肤结节病史30 余年,且呈加重趋势。有高血压病史30 余年,长期服用“苯磺酸氨氯地平”控制血压,血压控制不佳,最高达180/100 mmHg(1 mmHg=0.133 kPa)。既往黑便病史3 个月余。此外,患者的两个女儿皮肤上有多个结节,均已确诊NF1。查体:全身皮肤广泛肿瘤样肿物,质地软,表皮广泛分布咖啡斑,皮下瘀斑,眼底检查未见视神经萎缩、视乳头水肿及虹膜错构瘤结节(Lisch 结节)。血常规提示:白细胞3.6×109/L,红细胞3.54×1012/L,血红蛋白61.0 g/L,血小板308.0×109/L;肾上腺功能相关血液学结果:甲氧基肾上腺素(MN):0.96 nmol/L,甲氧基去甲肾上腺素(NMN):3.40 nmol/L;腹部CT 显示左肾上腺外侧支26 mm×25 mm 肿块软组织密度影,平扫39 HU,考虑左肾上腺PHEO 可能性大(图1A-B)。左上腹可见大小54 mm×35 mm 肿块状软组织密度影,考虑SIST 可能性大(图1C)。胸部、腹部、背部及双侧大腿皮下多发团块状软组织密度影,较大者约45 mm×23 mm(图1D)。电子胃肠镜检查无明显异常。完善相关检验检查后,考虑到患者黑便病史,且合并重度贫血,结合患者影像学检查,初步诊断为SIST。患者存在进行性消化道出血风险,此次手术以根治为主。患者术前血压控制不佳,不宜同时行肾上腺肿物切除术,综合胃肠外科及泌尿外科会诊意见后决定先行腹腔肿瘤切除术,术后药物调控血压1 个月后再行肾上腺肿瘤切除。手术顺利,术中快速病检回报:梭形细胞瘤(间质瘤可能性大)。空肠切除的小肠肿物大小6 cm×4.5 cm×2.5 cm,切除肿物HE 染色提示肿瘤包膜钙化,肿物内含出血、坏死及胆固醇裂隙区,病检结果回报:SIST(高度侵袭危险性),未见明确脉管内瘤栓及神经周侵犯。免疫组化结果显示:S-100(-)、SOX10(-)、Ki-67(2%+)、CD34(部分+)、CD117(+)、Dog-1(+)、desmin(-)、SMA(-)、SDHB(+)、STAT6(-)、nestin(+)、FH(+)、CK-Pan(-)、vimentin(+)、HHF35(-)(图1E)。基因检测结果回报:PIK3CA p.H1047R突变,未检测到KIT、PDGFRA 突变。同时,笔者对患者皮肤肿块进行了基因检测,结果显示:NF1c.586+1G>a 突变,符合NF1(图1F-G)。患者恢复顺利,于术后第5 天出院。术后3 个月,患者在我院泌尿外科行肾上腺肿块切除术,切除的肾上腺肿瘤呈灰红色,大小为3 cm×4 cm×3 cm,与正常肾上腺组织相连,包膜完整,血供丰富,术中血压波动大。对于组织病理学检查,标本用10%福尔马林固定,石蜡包埋并用HE 染色。腹部和躯干皮下结节的HE 染色表明,肿瘤由松散的梭形细胞组成,细胞质少、染色浅,细胞核薄、呈波浪状,细胞包埋在纤维样基质中。病理检查提示:PHEO(左肾上腺内、外侧肢)。免疫组化结果显示:S-100(-)、突触素(+)、嗜铬粒蛋白A(++)、神经元特异性烯醇化酶(-)、CK7(-)、癌胚抗原(-)、Ki-67(-)、p53(-)(图1H)。结合临床和病理结果,患者诊断为NF1 合并巨大肾上腺PHEO 和SIST。术后随访1 年,血压平稳,无腹痛及消化道出血,术后3 个月复查腹部CT 无异常。已从患者处获得发布个人和医疗信息的书面知情同意书。
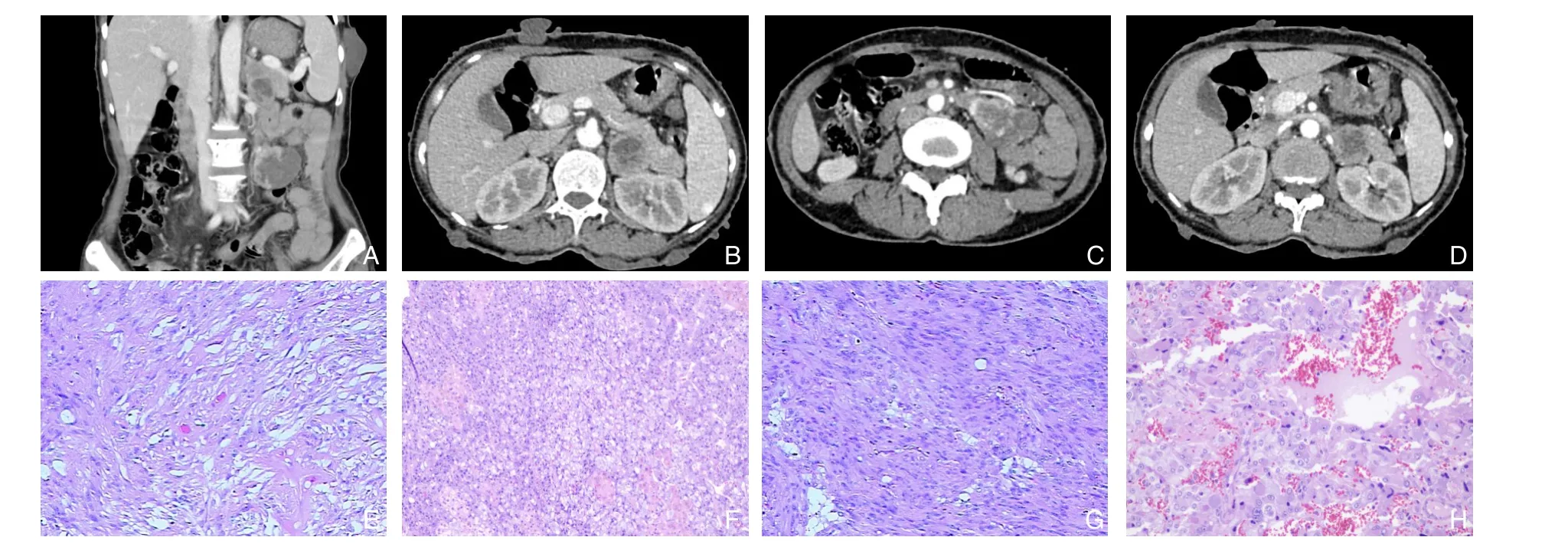
图1 影像学及病理学资料 A:患者肾上腺PHEO及SIST矢状位;B:患者PHEO横断位;C:患者SIST横断位;D:患者腹部皮下多发NF1;E:SIST组织形态学特征(HE×100):小肠肌层内可见大量梭形细胞束状排列,细胞核周常见空泡;F:NF1组织形态学特征(HE×100):可见排列密集的梭形细胞,呈波浪状,核细长,镶嵌在纤维基质中;G:NF1免疫组化(HE×200):绝大多数NF1细胞S-100(+),胞质、胞膜着棕黄色;H:PHEO组织形态学特征(HE×100):肿瘤细胞为多角形,胞质丰富,核仁明显,细胞排列呈巢状或腺泡状
2 讨论并文献复习
NF1 是一种与神经系统和其他器官系统畸形和肿瘤生长相关的遗传性肿瘤综合征,为常染色体显性遗传病。其最显著的临床表现为其皮肤病变,例如多个扁平、浅棕色的皮肤色素斑块(咖啡牛奶斑)、皮褶雀斑、皮下可见的神经纤维瘤和虹膜错构瘤结节(Lisch 结节)。由于在染色体17q11.2上发生抑癌基因NF1 突变,NF1 患者在某些肿瘤中的患病率较常人更高。0.1%~5.7%的患者合并肾上腺PHEO,8.8%合并胃肠道间质瘤,同时合并3 种疾病的病例极为罕见[5-10]。本研究中的病例表皮分布有广泛咖啡斑,有家族史,虽未发现丛状神经纤维瘤和Lisch 结节,但患者临床病理特征符合国际NF1 诊断标准诊疗指南[2]。
近年来,神经纤维瘤病合并PHEO 和胃肠道间质瘤的相关病例报告较少。笔者在PubMed 数据库中对相关文章进行了详细的文献检索。为尽可能避免文献缺失,笔者对检索获得的所有文章的参考文献也进行了检索,结果如表1 所示。除本研究报告患者外,全球仅发表了14 例涉及NF1 患者合并GIST 和PHEO 的类似病例。其中5 例发生于双侧肾上腺;13 例报告GIST 的位置,其中9 例位于小肠,5 例为恶性;14 例报告临床特征,其中10 例患者合并有高血压病史,2 例患者合并有腹痛,1 例患者合并有胃肠道出血,1 例患者合并有复发性镜下血尿。
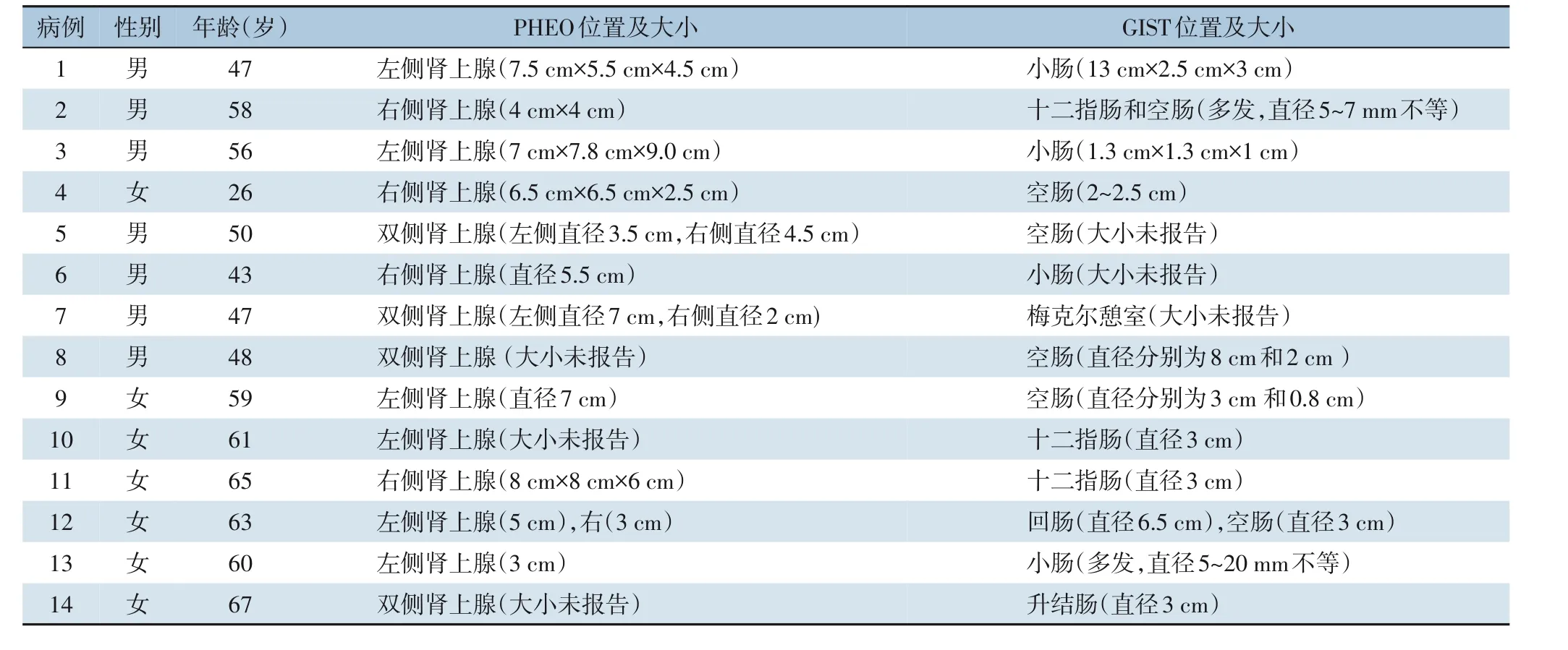
表1 全球范围内NF1患者合并PHEO及GIST的病例报道
GIST 是NF1 患者最常见的腹部肿瘤之一,其被认为起源于胃肠道肌层的Cajal 间质细胞。GIST不分良性或恶性,而是根据其恶性肿瘤的临床风险进行分层(极低、低、中等或高),约10%~30%的GIST 具有恶性临床病程[22]。GIST 的临床表现无特异性,早期患者多无临床症状或症状不典型,当肿瘤体积增大至压迫肠管或侵犯肠腔时可出现相应腹部症状,表现为消化道出血、腹痛、腹部包块、梗阻,甚至穿孔,本研究中患者因消化道出血症状入院。与NF1 相关的GIST 多发于小肠远端[23-24]。大约80%的GIST 中存在KIT 基因突变。细胞内膜结构域的功能获得性突变、缺失或插入缺失(由KIT 外显子11 编码)是KIT 中最常见的突变,可破坏KIT 基因的正常自身抑制状态。大约15%的GIST 是由PDGFRA 基因激活驱动的,显示KIT 受体同源区域的致癌突变(外显子12、14 和18)[25-27],野生型GIST 占所有GIST 中的5%~10%,不携带KIT 或PDGFRA 突变。然而,笔者总结近20 年相关病例报告,其中在NF1 相关GIST 中很少报告KIT 和PDGFRA 突变。NF1 相关GIST 总体恶性程度较低,且常对伊马替尼耐药,通常不需要药物治疗[28]。NF1 相关GIST 和野生型GIST 之间可能存在共同途径,即NF1 基因突变破坏神经纤维蛋白的正常功能并激活结构RAS,从而通过丝裂原活化蛋白激酶(MAPK)途径增加下游信号传导[29]。RAS-MAPK 级联反应的激活也可能由KIT 的突变引起[30],这种共同途径的激活导致GIST 的发展[31]。
本研究中患者的SIST 标本基因检测结果提示PIK3CA p.H1047R 突变。PIK3CA 编码磷脂酰肌醇-3-激酶(PI3K)的p110α 催化亚基,PI3K 是一种脂质激酶,是KIT 信号通路中促进细胞生长、增殖的下游效应因子。PI3K/Akt/mTOR 信号通路的激活在不同类型的人类癌症中均有报道,且常由PIK3CA 突变引起[21]。研究[32-34]表明,这种突变对于选择靶向药物治疗至关重要,酪氨酸激酶抑制剂可能对存在PIK3CA 突变的GIST 患者无效,对于此类患者,首选治疗应是PI3K/Akt/mTOR 通路的抑制剂。PIK3CA 的突变的存在,可能成为未来对NF1-GIST 进行精准治疗的关键靶点。
GIST 的药物治疗首选伊马替尼。它是晚期GIST 的一线治疗药物,也用于局部GIST 的新辅助和辅助治疗[24]。伊马替尼是一种能够抑制KIT、PDGFRA 和BCR/ABL 的新型酪氨酸激酶抑制剂(TKI),对伊马替尼敏感且具有高复发风险的KIT或PDGFRA 突变的GIST 患者在伊马替尼辅助治疗后生存期延长,在中位随访时间10.9 年时显示总缓解率超过50%,中位无进展生存期为1.7 年[35]。对于NF1-GIST,欧洲肿瘤内科学会指南[36]建议不要常规给予伊马替尼治疗,因为NF1-GIST 患者通常缺乏KIT 或PDGFRA 突变。因此,对于此类患者,进行基因检测以指导用药是很有必要的。对于伊马替尼治疗无效或对伊马替尼不耐受的患者,舒尼替尼和瑞戈非尼可分别作为二线和三线治疗[37-38]。
对于可切除非转移性GIST,进行完整R0切除手术是其主要治疗方法;1~2 cm 的肉眼可见切缘可能足以实现显微镜下阴性边缘。不同于消化道恶性肿瘤的手术要求,GIST 手术通常不需要进行淋巴结清扫,因为在局部GIST 中淋巴结受累的情况罕见[36,39]。
PHEO 是NF1 的另一种罕见腹腔肿瘤,大约在0.1%~5.7%的NF1 患者中发现了PHEO[40-41]。近期的一项研究[42]表明,0.3%(1/342)的NF1 患者合并PHEO。相对于正常人群,NF1 患者的PHEO 发病率更高,NF1 伴高血压患者的肾上腺良性PHEO 发生率可高达50%[43]。除此之外,84%的NF1相关PHEO表现为单发,且极少发生于肾上腺外(6.1%)[44]。PHEO 的发生可能与NF1 基因突变导致神经纤维蛋白合成缺失有关。Gutmann 等[45]的研究证实6 例NF1 患者的PHEO 病理切片中缺乏神经纤维蛋白表达,这项研究支持神经纤维蛋白可能参与NF1 相关PHEO 发病机制的观点。另外一项研究[46]表明,在NF1 相关PHEO 中进行的基因表达和生物学途径分析显示多种参与细胞分化,细胞黏附和运动以及细胞和器官发育过程的基因的激活,这可能是NF1 患者中观察到的PHEO 恶性程度稍高的原因。PHEO 的典型症状为儿茶酚胺分泌过多所致的阵发性或持续性高血压,CT 检查和儿茶酚胺中间代谢产物甲氧基肾上腺素(MN)和甲氧基去甲肾上腺素(NMN)检测分别是PHEO 定位和定性诊断方法。本例患者有多年阵发性高血压病史,术前专科检验结果提示MN 为0.96 nmol/L,NMN 为3.40 nmol/L。对中老年NF1 患者,如有反复高儿茶酚胺血症,应考虑PHEO 的可能,对此类患者,应测定血浆和尿液儿茶酚胺及代谢产物,以确定激素代谢水平。同时,肾上腺PHEO 的CT 和MRI 影像学表现具有一定的特征性,并能很好地显示肿瘤的部位、形态、大小、边界及邻近组织的关系,结合临床表现可明确诊断。
对于非转移性PHEO,手术切除是最佳治疗方案[47-48]。所有具有激素功能的PHEO 患者均应进行术前治疗,以预防围手术期心血管并发症,肾上腺素能受体阻滞剂是首选[49-50]。术前药物治疗应至少持续7~14 d,从而留出充足的时间使血压和心率恢复正常。同时,在手术后应密切监测血压、心率和血糖水平24~48 h[51-52]。在手术方式选择上,目前多采用腹腔镜下肾上腺切除术治疗大多数肾上腺PHEO。由于肾上腺PHEO 具有很高的复发和转移风险,在术式上首选全肾上腺切除术[51,53]。同时指南[54]建议对较大(>6 cm)或侵袭性PHEO 行开腹切除,以确保肿瘤完全切除,防止肿瘤破裂,避免局部复发。术中应密切监测血压变化,血压下降不明显或下降后迅速升高的患者应警惕肿瘤残留或转移灶的存在。术后应终身监测PHEO 复发情况[54-55]。
综上所述,NF1 合并GIST 和PHEO 的病例临床罕见,全面的血液学检查和影像学检查是明确诊断的关键。
利益冲突:所有作者均声明不存在利益冲突。
作者贡献声明:郑林熠及李玉强负责设计文章的思路、病例收集及文章的撰写;所有作者对文章进行书写及校对。
