应用生物信息学筛选结直肠癌Hub基因及验证
2024-04-24陈树华温日葵祝惠钦谢荣章
陈树华,温日葵,祝惠钦,谢荣章
云浮市人民医院检验科,广东云浮 527300
结直肠癌为临床中常见的消化道恶性肿瘤之一,其病死率在癌症中排第3位,严重威胁人类健康。因此,早期识别结直肠癌诊断和预后相关生物标志物至关重要[1]。生物信息学为生物学、信息学相结合的内容,在阐述疾病分子机制方面起着积极的作用。基因芯片主要是通过微阵列技术将高密度DNA片段阵列依附着玻璃、尼龙等材料上,筛选出有价值的基因进一步研究分析,目前常用于收集疾病表达谱数据[2]。Hub基因是经生物信息学筛选出的核心基因,也是当前临床疾病治疗的潜在靶点[3]。云浮市人民医院检验科运用生物信息学方法,筛选出结直肠癌相关的差异表达基因予以生物通路富集分析、制作蛋白-蛋白互作网络,筛选出Hub基因,并于2017—2022年8月收集30例结直肠癌组织和30例正常结直肠组织样本进一步验证Hub基因表达,为今后临床诊治结直肠癌、评估预后转归提供新型的辅助工具。现报道如下。
1 对象与方法
1.1 研究对象及芯片数据资料
收集2017—2022年8月本院就诊的30例结直肠癌患者组织标本作为异常组,另收集30例非癌结肠组织作为正常组。从美国国立生物技术信息中心的基因表达综合数据库(Gene Expression Omnibus, GEO)中下载结直肠癌芯片数据GSE 21815、GSE 31905、GSE 35279资料,其中GSE 21815包括12例结直肠癌组织和9例正常结直肠对照组织;GSE 31905包括55例结直肠癌组织和7例正常结直肠组织;GSE 35279包括74例癌组织和5例正常组织。
1.2 方法
1.2.1 数据预处理 运用tidyverse软件对获取的芯片数据资料予以数据处理,对各样本的基因文件进行命名,并把基因的表达量置于同一表达矩阵内。在metadata元数据库中把样本注释内容提取出来,使其与表达矩阵相符。最后通过R语言的biomaRt包对标准基因符号进行标注,进而取得标准化的基因表达矩阵。
1.2.2 筛选差异基因 应用edgeR包、limma包将数据预处理获得的基因表达矩阵进行差异表达基因分析,再应用GEO中的分析工具analyze with geo2r分为结直肠癌组织组和正常组织组,以∣logFC∣>2,P<0.01作为有效基因的纳入标准[4],筛选出GSE 21815、31905、35279数据集中的差异基因,并绘制韦恩图,找出GSE 21815、31905、35279数据集中的共有基因。
1.2.3 提取lncRNA 在Gencode数据库中收集和整理经过筛选的差异基因lncRNA相关信息,通过R软件lncRNA内ensembl ID及其表达量等信息。
1.2.4 生物功能富集分析 运用生物信息学工具DAVID,将与正常结直肠组织有差异的结直肠癌相关差异表达基因(Differentially Expressed Genes,DEGs)筛选出来,通过基因本体(Gene Ontology,GO)开展功能富集分析(包括细胞组分、分子功能、生物过程3个部分),并进行京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析。
1.2.5 蛋白互作网络(Protein Protein Interaction,PPI)分析 在STRING数据库上参与富集分析的DEGs基因进行PPI分析,应用Cytoscape构建PPI网络。具体操作为:借助网络分析算出网络中心性参数—度中心性,用MCODE插件提取关键子网,获得相对集中的表达子集,识别出结直肠癌的潜在调控基因。最后,用ClueGO、CluePedia把KEGG通路可视化,绘制生物通路之间的基因互作网络,提取与通路之间相互作用程度排名前10的差异基因作为Hub基因。
1.2.6 结直肠癌组织样本Hub基因表达验证 应用实时荧光定量PCR(Quantitative Real-Time PCR,qPCR)测量癌组织、正常结直肠组织中hub基因表达量没具体方法。①提取总RNA:取0.3~0.5 g组织样本,将其研磨成粉末状,置于1.5 mL离心管内,根据RNA提取试剂盒说明书流程提取总RNA。②逆转录反应:根据逆转录试剂盒的操作流程进行逆转录反应,取200 ng RNA量,滴加1 μl特异性逆转录引物+12 μL无菌水,孵育5 min后,依次滴加Ribollock RNase抑制剂、RevertAid M-MuLV反转录酶。③RT-PCR反应检测:以SYBR green法,设置反应体系:95℃ 10 min,95℃ 15 s,60℃ 1 min,40个循环,以U6作为内参基因,以2-△△Ct作为基因表达量,以正常结直肠组织为对照,得出结直肠癌组织中hub表达量。△△Ct=(Ct目的基因-CtU6)结直肠癌组织-(Ct目的基因-CtU6)正常结直肠组织。
1.3 统计方法
应用SPSS 26.0统计学软件进行数据统计分析,计量资料(mRNA表达量)符合正态分布,以()描述,行t检验,P<O.05为差异有统计学意义。
2 结果
2.1 差异基因筛选结果
GSE 21815(937)、31905(1470)、35279(1458)数据集共有141例结直肠癌组织和21例正常结直肠组织,3个数据集分别检索到937个、1 470个、1 458个DEGs基因,在软件上绘制韦恩图后获得共有上调基因105个、共有下调基因140个。
2.2 核心基因验证
经PPI网络图分析可知,245个共有差异基因导入到STRING数据库生成PPI网络图,并对PPI网络图进行可视化分析得出与PPI网络相互作用程度最高的10个Hub基因,分别为催产素受体基因(Oxytocin Receptor, OXTR)、基质金属蛋白酶11基因(Matrix Metalloproteinase-11, MMP11)、酪氨酸蛋白激酶受体B2基因(Erythropoietin-Producing Hepatoma-B2, EPHB2)、间质上皮转化因子基因(Mesenchymal-Epithelial Transition Factor, MET)、转化生长因子β1基因(transforming Growth Factor-β1, TGF-β1)、抑制斯钙素-2基因(Stanniocalcin-2, STC2)、基质金属蛋白酶7基因(Matrix Metalloproteinase-7, MMP7)、激肽释放酶8基因(Kallikrein-8, KLK8)、激肽释放酶10基因(Kallikrein-10, KLK10)、角蛋白23基因(Keratin-23, KRT23)。结直肠癌组织的MMP11、c-MET、MMP7、KLK8、KLK10 mRNA表达量明细高于正常结直肠组织,差异有统计学意义(P均<0.05)。而结直肠癌组织与正常结直肠组织的OXTR、EPHB2、TGF-β1、STC2、KRT23 mRNA表达比较,差异无统计学意义(P均>0.05),见表1。
表1 结直肠癌组织与正常结直肠组织10个Hub基因mRNA表达量比较()
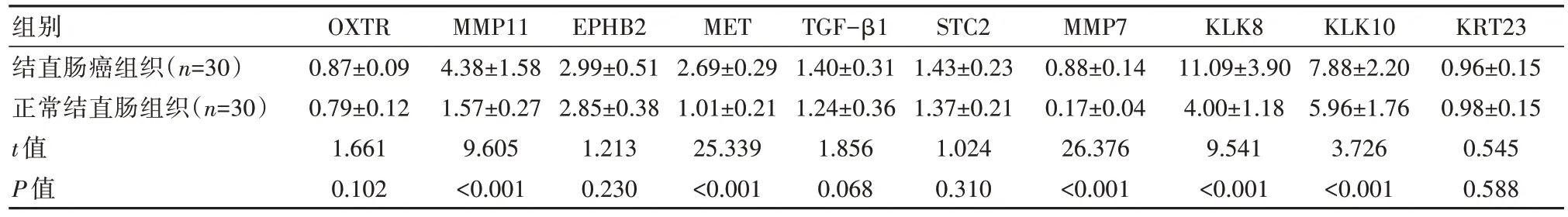
表1 结直肠癌组织与正常结直肠组织10个Hub基因mRNA表达量比较()
注:OXTR:催产素受体基因,MMP11:基质金属蛋白酶11基因,EPHB2:酪氨酸蛋白激酶受体B2基因,MET:间质上皮转化因子基因,TGF-β1:转化生长因子β1基因,STC2;抑制斯钙素-2基因,MMP7:基质金属蛋白酶7基因,KLK8:激肽释放酶8基因,KLK10:激肽释放酶10基因,KRT23:角蛋白23基因。
KRT23 0.96±0.15 0.98±0.15 0.545 0.588组别结直肠癌组织(n=30)正常结直肠组织(n=30)t值P值OXTR 0.87±0.09 0.79±0.12 1.661 0.102 MMP11 4.38±1.58 1.57±0.27 9.605<0.001 EPHB2 2.99±0.51 2.85±0.38 1.213 0.230 MET 2.69±0.29 1.01±0.21 25.339<0.001 TGF-β1 1.40±0.31 1.24±0.36 1.856 0.068 STC2 1.43±0.23 1.37±0.21 1.024 0.310 MMP7 0.88±0.14 0.17±0.04 26.376<0.001 KLK8 11.09±3.90 4.00±1.18 9.541<0.001 KLK10 7.88±2.20 5.96±1.76 3.726<0.001
3 讨论
目前已有大量基础研究、临床研究揭示了结直肠癌发生发展的病因病机[5-6],但其患病率、病死率近年仍居高不下,有学者认为其原因主要是大多数研究是针对单个遗传学事件进行队列研究,造成结果偏倚[7]。本研究选择GSE 21815、GSE 31905、GSE 352793个数据库运用生物信息学深入研究,筛选获得245个结直肠癌相关差异表达基因,而且对上调、下调245个共有差异基因进行GO生物功能分析结果显示,①上调DEGs基因的细胞组分(Cellular Component, CC)涉及细胞外基质、细胞外区域、细胞质膜等;下调基因细胞组分涉及细胞外区域、细胞外谜题、细胞间膜蛋白等。②上调基因的分子功能主要是序列特异性DNA结合蛋白活性、RNA聚合酶Ⅱ、生长因子激活等;下调基因分子功能主要为激素激活体系、结合锌离子的蛋白质。③上调基因的生物过程(Biological Process, BP)包括细胞增殖、细胞凋亡、药物反应、蛋白质水解等;下调基因生物过程包括蛋白质水解、小分子物质跨膜转运等。经KEGG富集分析可知,上调DEGs基因主要富集分布转化生长因子-β(Transforming Growth Factor-β, TGF-β)信号通路、Wnt信号通路;下调DEGs基因富集于PPAR信号通路、氮代谢信号通路等。由此可见,上调基因主要分布在细胞核、细胞膜、细胞外,通过调节DNA复制、参与细胞周期等过程以参与结直肠癌细胞的细胞迁移、增殖和凋亡。而下调基因主要分布在细胞外区域,参与机体代谢,进而对癌症发生发展起作用。
本研究制作差异基因蛋白互作PPI网络获得10个Hub基因,并经临床组织样本验证发现,结直肠癌组织中基质金属蛋白酶11基因(4.38±1.58)、间质上皮转化因子基因(2.69±0.29)、基质金属蛋白酶7基因(0.878±0.143)、激肽释放酶8基因(11.09±3.90)、激肽释放酶10基因mRNA(7.88±2.20)mRNA表达显著高于正常结直肠组织组织(P均<0.05)。基质金属蛋白酶(MMPs)对细胞外基质、基底膜蛋白酶有溶解作用,在正常情况下该物质在体内的表达水平较低,但若发生病理改变,特别是出现癌细胞增殖分化、迁移等,其表达水平会显著升高[8]。MMP11就是MMPs中的一员,可通过重构细胞外基质诱导肿瘤进展,并可通过抑制细胞凋亡使肿瘤细胞存活[9]。MMP7是MMPs中分子量最少的分泌蛋白,其能够溶解细胞外基质,并可水解机体蛋白多糖、胶原底物,促进肿瘤细胞增殖分化[10]。目前已有研究报道,MMP11(4.25±1.19)、MMP7(0.886±0.125)在多种癌组织中呈高表达[11]。李军彦等[12]的研究显示,MMP7表达能够提示结直肠癌病情发生及其进展。温凌等[13]研究也发现,检测MMP-11能够辅助诊断结直肠癌发生发展情况。C-Met为原癌基因的一种,能够使活化信号传导通路发生磷酸化,促使细胞运动,进而引起上皮细胞分散、内皮细胞迁移,肿瘤细胞发生浸润迁移。另有研究报道,C-Met能够诱导肿瘤血管新生[14]。曹明等[15]研究证实,结直肠癌患者癌组织CMet表达水平与癌细胞侵袭转移呈正相关,对于判断预后有一定指导意义。已有研究证实,KLKs与恶性肿瘤发生发展密切相关,其作用机理是通过编码hK蛋白溶解肿瘤细胞外基质蛋白,促使癌细胞迁移和肿瘤血管新生[16]。相关文献报道,KLK8能够促进结直肠癌细胞增殖、转移,其过表达能够抑制肿瘤细胞凋亡,敲低KLK8后会促使癌细胞凋亡[17]。另有研究发现,KLK10在结直肠癌组织中异常表达,与肿瘤病理分期、肝转移发生呈正相关[18]。由此可见,MMP11、c-MET、MMP7、KLK8、KLK10高表达,能够促进结直肠癌细胞增殖分化、浸润迁移、
综上所述,本文利用生物学信息方法进行筛选和验证了一组与结直肠癌发生发展密切相关的基因,有望成为作为早期预测结直肠癌的标志物,为后续结直肠癌基础研究及临床诊疗提供依据。
