两型豆属叶绿体基因组特征及密码子偏好性分析
2024-04-11余潇赵振宁邓莉兰
余潇 赵振宁 邓莉兰

摘 要 为明确锈毛两型豆的叶绿体基因组结构和两型豆属叶绿体基因组密码子使用偏性及影响因素,以亚热带中、南部地区具有广阔开发利用前景的豆科草种—锈毛两型豆(Amphicarpaea ferruginea)为试验材料,利用高通量测序技術对锈毛两型豆进行叶绿体基因组测序、组装和注释,对其叶绿体基因组结构、基因组成进行分析。同时利用 CodonW 1.4.2 软件和CUSP 在线程序等软件分析锈毛两型豆和两型豆的基因密码子使用偏性参数和核苷酸组成。结果显示:锈毛两型豆叶绿体基因组全长为 152 531 bp,包含83 364 bp的大单拷贝(LSC)区、17 935 bp的小单拷贝(SSC)区和25 616 bp的1对反向重复序列,为典型四分体结构,GC含量为35.44%;叶绿体基因组共编码130个基因,包括85个蛋白质编码基因、37个tRNA基因和8个rRNA基因;叶绿体基因组共检测出73个简单重复序列(SSRs),单、二、三、四、五和六核苷酸SSRs的数目分别为41、28、3、1、0和0。从锈毛两型豆和两型豆叶绿体基因组中筛选到适用于密码子使用偏好性分析的CDS基因共48条,两种植物叶绿体基因组具有相似的密码子使用模式,均倾向于使用A与U作为末尾碱基,48个基因的ENC 值均在35%以上,表明锈毛两型豆和两型豆叶绿体基因组的密码子偏性较弱;综合中性绘图分析、有效密码子数(ENC-plot)分析和奇偶偏好性(PR2-plot)分析的结果显示两型豆属叶绿体基因组密码子使用模式不仅受突变压力的影响,同时还受选择压力以及其他多种因素的影响;在锈毛两型豆和两型豆中分别确定18和20个最优密码子,其中共有最优密码子18个。基于豆科20个物种叶绿体基因组构建的系统发育树表明,豆科20个物种构成一个单系类群,锈毛两型豆与两型豆亲缘关系最近。
关键词 两型豆属;锈毛两型豆;叶绿体基因组特征;密码子偏好性;系统发育分析
两型豆属(Amphicarpaea Elliot)隶属于豆科,该属大多为多年生缠绕草本植物,全球共有约10种,广泛分布于东亚、北美以及非洲东南部等地,中国产3种,分别为两型豆(Amphicarpaea edgeworthii Benth.)、锈毛两型豆[Amphicarpaea ferruginea (Franch.) Y. T. Wei]和线苞两型豆(Amphicarpaea linearis Chun et T. Chen)[1]。两型豆属植物最显著的特征是在地上和地下均能发育出成熟果实,是典型的地上地下两型结实类型[2]。由于两型豆属植物这一独特的发育模式,其已成为植物发育生物学研究的模式植物,具有独特的育种意义[3]。两型豆属植物与大豆属植物的形态特征非常相似,在亲缘关系上也非常近,研究者们希望研究找到控制两型豆属植物发育方式的性状基因,使大豆也能实现地上和地下结果,以此提高豆类作物的产量[4]。此外,两型豆属植物的粗蛋白、钙和磷含量较高,具有较高的饲用价值,是优质牧草发展的重要对象;种子富含异黄酮类化合物,具抗炎、抗氧化、抗肿瘤、抗菌等作用[5]。锈毛两型豆[Amphicarpaea ferruginea(Franch.)Y.T.Wei]是两型豆属的多年生草本藤本植物,主要分布在中国云南省和四川省海拔2 300~3 000 m的山坡林中[6],为中国特有种,国家二级重点保护野生植物;而两型豆(Amphicarpaea edgeworthii Benth.)则为1 a生缠绕草本植物,在中国分布较广,常生于海拔 300~1 800 m的山坡、路旁及旷野草地,具较强的耐荫性,其营养价值与经济价值与锈毛两型豆相似[7]。目前对于两型豆和锈毛两型豆的研究主要集中在种子营养成分、种子休眠解除与解剖观察等方面[6-7]。锈毛两型豆和两型豆作为两型豆属植物的典型物种,《中国南方牧草志 第一卷 豆科》中记载其为亚热带中、南部地区具有广阔开发利用前景的豆科草种[8]。
叶绿体是重要的植物细胞器,在光合作用、生物合成和碳储存中起着至关重要的作用,具有独立于核基因组的遗传系统。自从烟草的第一个叶绿体基因组[9]问世以来,人们对其结构和功能的了解日益加深。叶绿体基因组的长度为100 ~200 kb,具有典型的四分体结构,包括大单拷贝区(LSC)、小单拷贝区(SSC)和两个反向重复区(IR)[10]。密码子使用偏好性是指编码相同氨基酸的同义密码子频率的差异,这种现象在原核生物和真核生物的生物体中都很常见[11]。然而,不同的基因组有其特有的同义密码子使用模式,这使得解释这种偏好性具有挑战性[12]。以往的研究主要集中于核基因组中密码子的使用偏性[13-14],而针对其细胞器基因组的研究相对较少。作为基因相对保守的叶绿体基因组使用的是不同于标准密码子的第十一套密码子表[15]。通常,密码子使用偏性反映了物种或基因的起源、进化和突变模式,并会对基因功能和蛋白质表达产生重大影响[16]。因此,分析叶绿体基因组中的密码子使用偏好将有助于了解密码子偏好选择的潜在分子机制以及相关物种的进化和环境适应,同时也能研究物种间的进化关系,对研究基因表达具有重要意义。
两型豆属植物具有巨大的科研及经济价值,但目前仅有山东师范大学生命科学学院Han Y.提交的两型豆叶绿体基因组序列公布[7],而未见关于两型豆属叶绿体基因组特征和密码子偏性研究的详细报道,本研究通过对锈毛两型豆的叶绿体全基因组进行测序、分析,并与近缘种两型豆进行比较,进一步分析两型豆属植物叶绿体基因组密码子碱基组成、密码子偏好性、最优密码子以及系统发育关系,结果可为两型豆属饲草植物叶绿体深入研究和应用奠定基础。
1 材料与方法
1.1 DNA 提取及叶绿体基因组测序注释
所用的锈毛两型豆的新鲜植物叶片采集于云南省玉龙县黄山镇南溪村(东经 100°8′59.93″,北纬26°46′8.02″,海拔3 103 m)的野生植株,共采集3株,每株上采集3片成熟叶片,经西南林业大学标本馆树木学教研室李双智副教授鉴定为豆科两型豆属植物锈毛两型豆Amphicarpaea ferruginea(Franch.)Y.T.We。使用改良的CTAB法[17]从硅胶干燥的叶片中提取总基因组DNA,提取的DNA送天津诺禾致源生物科技有限公司(中国天津)进行叶绿体基因组测序,使用Get organelle组装叶绿体基因组,生成的完整叶绿体基因组的组装图由Bandage v.0.8.1验证[18]。使用PGA软件,以两型豆(Amphicarpaea edgeworthii)叶绿体基因组序列为参考,对序列进行注释,然后在Geneious Prime 10.0.5中手动调整[19]。利用Organellar Genome DRAW在线程序生成注释的叶绿体基因组图,锈毛两型豆的完整叶绿体基因组提交至GenBank公共数据库,登录号为ON050971。从NCBI数据库(https://www.ncbi.nlm.nih.gov/)平台上下载两型豆的叶绿体基因组,登录号为NC_057598.1。
1.2 重复序列分析
采用在线工具MISA-web(http://webblast.ipk-gatersleben.de/misa/)对锈毛两型豆的SSRs的类型和数量进行分析[20],对单、二、三、四、五和六核苷酸的关键参数分别设置为10、5、4、3、3和3,两个SSRs之间的最小间距为100 bp。
1.3 密码子偏好性分析
1.3.1 密码子相关参数计算 根据GenBank文件中的信息手动提取每个叶绿体基因组中的蛋白质编码序列,然后检查其是否存在正确的起始密码子和终止密码子。由于短长度的CDS通常会导致密码子使用的较大估计误差,因此在密码子使用计算中,长度小于300 bp的CDS被排除在外,以避免采样偏差[21]。利用CUSP在线程序(http://www.Bioin formatics.nl/emboss-explorer/)计算整个基因的GC含量(GCall)、第1、第2和第3位密码子位置(GC1、GC2、GC3)。
1.3.2 中性绘图分析 GC12是GC1和GC2的平均值,手动计算用于中性图分析。中性分析(GC12 vs GC3)是GC12与GC3的对比图,有助于揭示GC12和GC3之间的关系,用于检查密码子形成过程中的突变选择平衡[22]。在中性图中,单个基因由一个离散点表示,如果GC12与GC3为中性,则这些点应位于对角线上(单位斜率),而如果GC12不为中性,这些点应显示在横坐标的平行线上(零斜率),斜率小于1的程度表明GC12的中性程度小于GC3[23]。
1.3.3 ENC-plot分析 ENC图分析(ENC vs GC3)用于检查基因组的密码子使用是否仅受突变力或其他因素的影响,遵循的标准是,对于一个基因,如果其密码子使用仅受G+C突变偏倚的限制,则其将位于或略低于预测值的曲线,而如果要选择翻译最佳密码子,它将大大低于预期曲线[24]。根据各组基因密码子的GC含量和ENC值,运用R语言绘制ENC-plot散点图,并计算出预期ENC值(预期ENC计算公式:ENCexp= GC3+2+29/[GC23+(1-GC3)2]),通过比较预期ENC值与实际ENC值,可以分析得出突变压力和选择压力对密码子使用偏性的作用大小[25]。单纯分析ENC-plot绘图无法定量比较实际ENC值与标准ENC值的差异,因此通过计算得到ENC比值(即(标准ENC-实际ENC)/标准ENC),并统计各个组段ENC比值的频数和频率,从而更加准确直观地对其ENC差异进行分析。
1.3.4 PR2-plot绘图 奇偶校验分析(PR2)是DNA组成的规则,用于指示突变和选择压力对密码子使用偏差的影响。分析每个密码子第3位的A、T、C和G的含量,并以A3/(A3+T3)为纵坐标,以G3/(G3+C3)为横坐标进行PR2-plot偏差图分析。每个基因的碱基组成显示在一个平面上,其中中心点表示无偏使用时的密码子状态,即A=T和C=G,其余点和中心点之间的矢量距离表示偏误的程度和方向[26]。
1.3.5 最优密码子确定 最优密码子被定义为在高表达基因中比在低表达基因中更频繁出现的密码子。以ENC为首选标准,将48条叶绿体基因中ENC值最高和最低的5个基因分别视为高表达组和低表达组。如果ΔRSCU>0.08,并且密码子的同义密码子的相对使用频率(RSCU)在高样本组大于1,在低样本组小于1,则定义为高表达最优密码子(高级密码子)[27]。
1.4 系统发育分析
以锈毛两型豆的叶绿体基因组序列在NCBI数据库上进行BLAST同源性比对,下载同源性比对率高于85%的序列,共计19个近缘物种。同时以蔷薇科的缫丝花Rosa roxburghii(NC_032038.1)和金樱子Rosa laevigata(NC_046824.1)两种植物为外类群,对锈毛两型豆及其19个近缘种构建系统发育树。首先,对所有这22个完整的叶绿体基因组序列均通过MAFFT v.7软件进行比对[28],基于RAxMLv.8中的GTR+ I+G模型,采用最大似然法进行系统发育分析[29],共设1 000次自展值重复。
1.5 数据统计
基于皮尔逊相关法,采用SPSS 18.0和Excel 2019软件进行相关分析,显著性水平为P<0.05或 P<0.01。
2 结果与分析
2.1 锈毛两型豆叶绿体基因组的基本特征
锈毛两型豆的叶绿体基因组是一种双链环状DNA,长度为152 531 bp,包含83 364 bp的大单拷贝(LSC)区、17 935 bp的小单拷贝(SSC)区和2 561 6bp的1对反向重复序列(图1)。叶绿体基因组的碱基组成不均匀,A碱基占32.26%、C碱基占17.60%、G碱基占17.85%、T碱基占32.29%,整个叶绿体组的GC和AT含量分别为35.45%和64.55%。IR区的GC含量(41.87%)高于LSC区(32.92%)和SSC区(28.81%)(表1)。葉绿体基因组共编码130个基因,包括85个蛋白质编码基因、37个tRNA基因和8个rRNA基因。18个基因在IR区复制并相互反向重复,包括7个蛋白质编码基因(rpl2、rpl23、ycf2、ndhB、rps7、rps12、ycf1)、7个tRNA基因(trnN-GUU、trnI-CAU、trnL-CAA、trnA-UGC、trnV-GAC、trnI-GAU、trnR-ACG)和4个rRNA基因(rrn4.5、rrn5、rrn16、rrn23)。从基因功能上看,与自我复制相关的基因有69个,与光合作用相关的基因 49个,另有 6 个其他编码蛋白质的基因和6个功能未知的基因 (表2)。
2.2 锈毛两型豆重复序列分析
利用在线软件MISA web在锈毛两型豆中共检测出73个SSRs(表3),单、二、三、四、五和六核苷酸SSRs的数目分别为41、28、3、1、0和0。其中,单核苷酸重复序列共有A(23个SSRs)、G (1个SSRs)和T(17个SSRs)3种重复类型;二核苷酸重复序列共有AT(17个SSRs)和TA(11个SSRs)2种重复类型;三核苷酸共有ATA(1个SSRs)、ATT(1个SSRs)和TCT(1个SSRs)3种重复类型;四核苷酸共有只有TATC(1个SSRs)1种重复类型。在 73个SSRs中,有13个SSRs 位于基因的编码区(coding sequence,CDS);有60个SSRs位于基因的非编码区,其中9个 SSRs位于基因的内含子(intron)区域,51个 SSRs 位于基因间隔区 (intergenic spacer,IGS)(表4)。
2.3 两型豆属密码子碱基组成分析
去除叶绿体基因组长度小于300 bp的CDS序列,从锈毛两型豆和两型豆叶绿体基因组中筛选到适用于密码子使用偏好性分析的CDS基因共48条。锈毛两型豆和两型豆叶绿体基因组的GC含量见表5。锈毛两型豆的48个CDS基因GC1、GC2、GC3和GCall的平均GC含量分别为45.78%(30.8%~56.9%)、38.29%(26.4%~54.7%)、25.67%(18%~35.8%)和36.58%(26.8%~43.3%),而兩型豆的分别为45.75%(31.1%~56.5%)、38.24%(26%~54.7%)、25.6%(17.5%~35.8%)和36.52%(26.4%~ 43.3%)。统计发现GC含量在密码子的3个位置之间的分布不均,其中 GC1>GC2>GC3,平均 GC 含量均小于 50%,这说明锈毛两型豆和两型豆的叶绿体基因组倾向于使用A与U作为末尾碱基。锈毛两型豆和两型豆叶绿体基因组CDS基因的 ENC 值分别在35.83%(rps18)~ 56.95%(atpE)和35.83%(rps18)~52.01(ycf3),两个物种的48个基因的ENC 值均在35%以上,表明锈毛两型豆和两型豆叶绿体基因组的密码子偏性较弱。
运用SPSS软件对锈毛两型豆和两型豆叶绿体全基因组的GC1、GC2、GC3、GCall和ENC之间的相关性进行分析(表6),锈毛两型豆相关性达到极显著水平 (P<0.01)的参数有4组,分别为GC1与GC2、GC1与GCall、GC2与GCall、GC3与GCall,而两型豆相关性达到极显著水平(P< 0.01)的参数有5组,分别为GC1与GC2、GC1与GCall、GC2与GCall、GC3与GCall、GC1与ENC,表明锈毛两型豆和两型豆的密码子第1、2位碱基的成分最为接近,与第3位上的碱基具有一定的相似性,且两型豆的密码子第1位碱基构成会对密码子的使用偏好性具有极显著影响。锈毛两型豆和两型豆相关性呈显著水平的参数均为4组,锈毛两型豆的分别为GC2与GC3、GC1与GC3、GC1与ENC、GC3与ENC,两型豆的分别为GC2与GC3、GC1与GC3、GC3与ENC、GCall与ENC,表明锈毛两型豆的密码子第 1位和第3 位碱基组成,两型豆的密码子第3 位碱基组成和基因编码区总GC含量对密码子使用偏性有重要影响。
锈毛两型豆和两型豆各氨基酸密码子的 RSCU 分析显示(表7),锈毛两型豆和两型豆的RSCU 值相近,锈毛两型豆和两型豆RSCU>1 的密码子均有 11 个,其中有6个密码子以A作为结尾,有5个密码子以U作为结尾,没有以G作为末尾碱基的密码子,由此可知锈毛两型豆和两型豆偏好于使用 A、U 作为密码子的末尾碱基。而在RSCU<1 的密码子中除UGA和CUA以A结尾外,其余密码子均以 C 或 G 结尾,说明锈毛两型豆和两型豆以 C和G 结尾的密码子出现频率比较低,是非偏好密码子。
2.4 两型豆属密码子偏好性分析
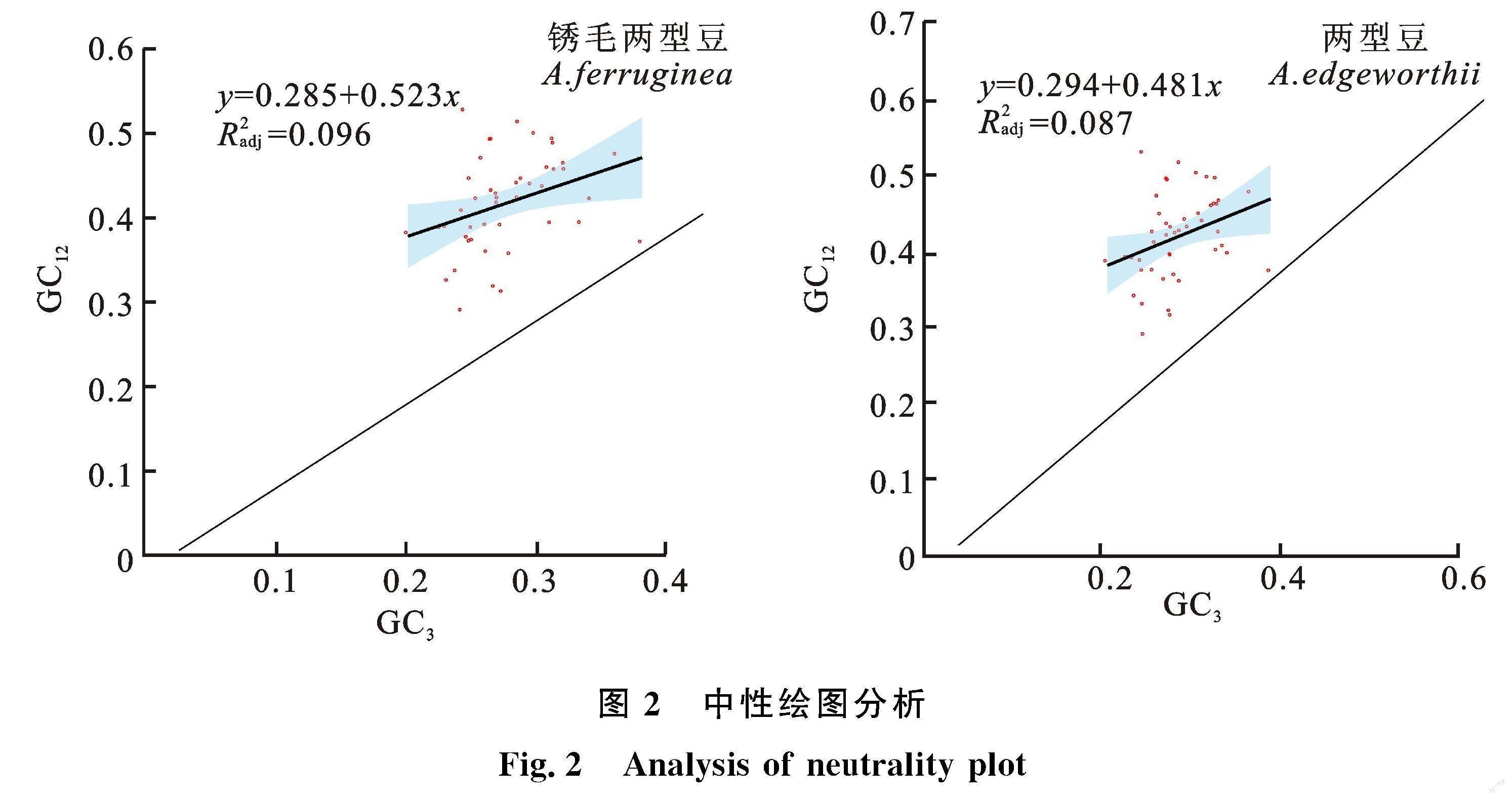
锈毛两型豆和两型豆中性绘图见图2。锈毛两型豆和两型豆叶绿体基因组各基因的GC3分别为18.0%~35.8%、17.5%~35.8%,GC3 含量最低的基因为 rps3基因,最高的为 ycf2基因。GC12的取值范围分别为锈毛两型豆(29.1%~52.9%)、两型豆(28.75%~52.90%)。锈毛两型豆和两型豆的GC12与 GC3 的相关系数分别为 0.096和 0.087,相关性均达到了显著水平(P< 0.05),说明突变压力对密码子偏好性的影响具有的显著的影响。锈毛两型豆和两型豆的 GC12与GC3回归系数分别为0.285和0.294,满足GC12和GC3呈显著相关性且回归系数大于0,说明锈毛两型豆和两型豆叶绿体基因组密码子碱基的第3位与第1、2位组成相似,密码子受突变压力的影响较大。
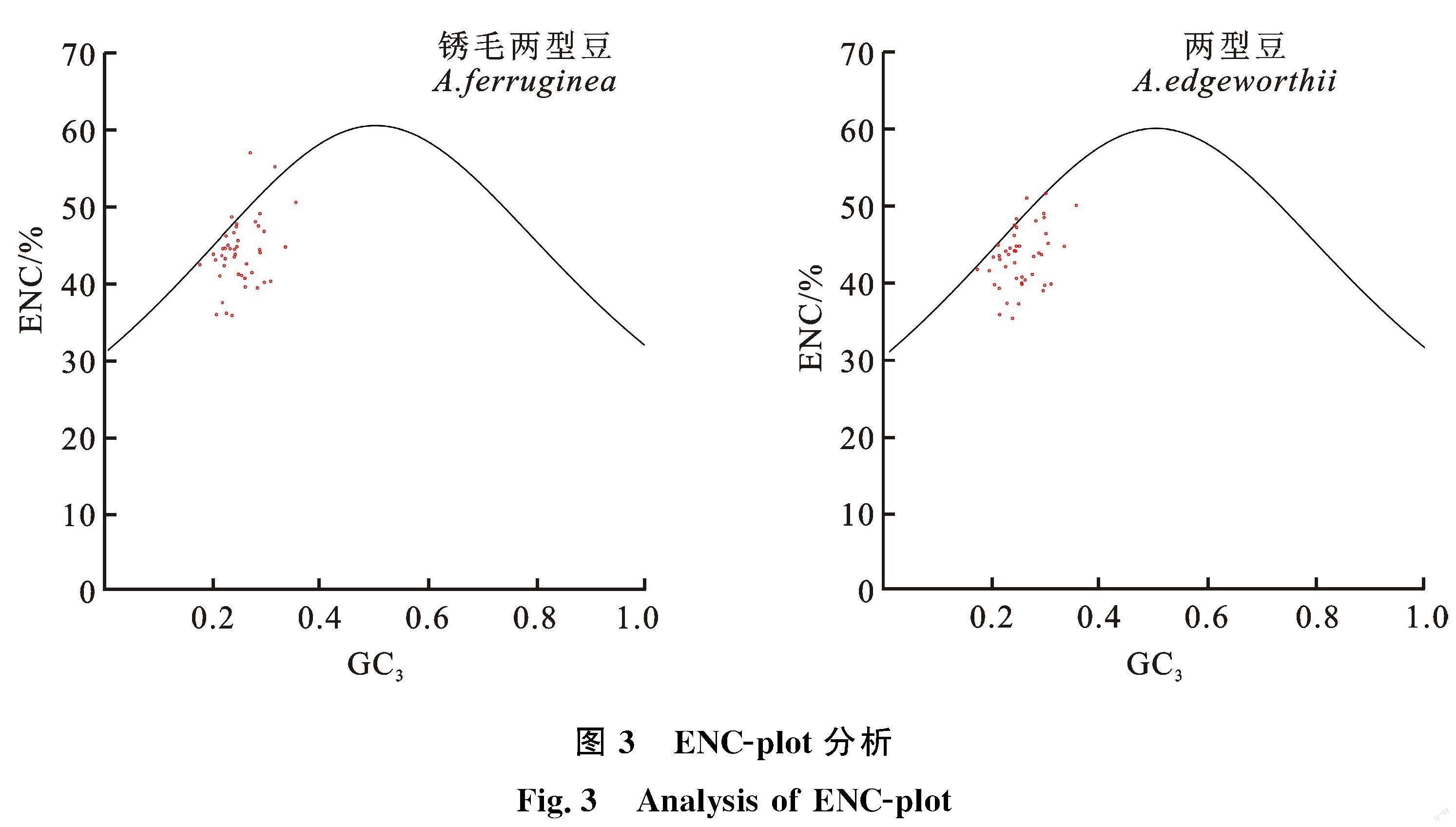
锈毛两型豆和两型豆叶绿体基因组密码子的ENC与 GC3的关联分析(图3)显示,分布在标准ENC曲线下方的基因较多,其大多与预期ENC值差距较大,仅有少数基因位于曲线附近,说明锈毛两型豆和两型豆叶绿体基因组密码子偏好性受选择压力的影响较大。进一步计算ENC比值,并对其ENC比值在各个组段的频数和频率进行分析(表8),锈毛两型豆和两型豆ENC比值分布在-0.05~0.05 区间的数量分别有 9个和10个,处于-0.05~0.05 区间外的基因分别为39个和38个,分别占到基因总数的81.25%和 79.16%,这一部分基因离标准曲线较远,表明锈毛两型豆和两型豆偏好性主要受到选择压力的影响,这与中性绘图分析的结果具有一定的差异。
采用 PR2-plot 绘图分析锈毛两型豆和两型豆各编码基因部分氨基酸嘌呤(A和G)和嘧啶(T和 C)之间的关系(图4),结果显示图中的大部分基因分布于右下角象限内,说明在碱基使用频率方面,密码子第3位T碱基的出现频率大于A碱基,G碱基的出现频率大于C碱基,嘧啶出现频率高于嘌呤,锈毛两型豆和两型豆叶绿体基因密码子的第 3 位碱基的使用存在T/C 偏向,其密码子使用模式不仅受突变压力的影响,同时还受选择压力以及其他多种因素的影响。[FL)]
2.5 两型豆属最优密码子的确定
按照48个CDS基因的ENC值对其进行排序,根据高低库中密码子的 RSCU 值和?傄cRSCU 值来确定锈毛两型豆和两型豆叶绿体的最优密码子(表 9)。锈毛两型豆高表达密码子(ΔRSCU≥0.08)有22个,高频密码子(RSCU>1)有30个,筛选得到GCU、AGA、CGA、AAU、UGU、CAA、GAA、GGU、CAU、AUA、UUA、AAA、UUU、CCU、AGU、ACU、UAU、GUU等共计18个最优密码子。而在两型豆中,表达密码子 (ΔRSCU≥0.08)有25个,高频密码子(RSCU≥1)有30个,筛选得到GCU、AGA、CGA、AAU、UGU、CAA、GAA、GGU、CAU、AUA、AUU、UUA、AAA、UUU、CCU、AGU、ACU、UAU、GUA、GUU等共计20个最优密码子。除AUU和GUA以外,其余均为两个物种的共有最优密码子,共计18个。18个共有密码子中 △RSCU均大于 0.5 的有 5个,分别为编码丙氨酸GCU、编码精氨酸AGA、编码精氨酸CGA、编码亮氨酸UUA和编码缬氨酸GUU。两个物种的最优密码子均以A或U结尾,共有最优密码子当中,以A结尾的最优密码子共计7个,以U结尾的有 11个。
2.6 系统发育分析
以缫丝花和金樱子为外类群,基于豆科20个物种叶绿体基因组构建的系统发育树表明,豆科20个物种构成一个单系类群,自展支持率(bootstrap)高达100%(图5),单系类群形成具有高支持率(100%)的 两大分支(Clade 1 和 Clade 2),其中Clade 1由小刀豆(Canavalia cathartica)、刀豆(Canavalia gladiata)、棉豆(Phaseolus lunatus)、四棱豆(Psophocarpus tetragonolobus)、美麗密花豆(Spatholobus pulcher)、密花豆(Spatholobus suberectus)黑吉豆(Vigna mungo)、蝶豆(Clitoria ternatea)、北美土圞儿(Apios americana)、大果油麻藤(Mucuna macrocarpa)、油麻藤(Mucuna sempervirens)、紫矿(Butea monosperma)、须弥葛(Haymondia wallichii)、豆薯(Pachyrhizus erosus)、锈毛两型豆(Amphicarpaea ferruginea)、两型豆(Amphicarpaea edgeworthii)、宽叶蔓豆(Glycine gracilis)和大豆(Glycine max)18个物种,Clade 2由山葛(Pueraria montana)和食用葛(Pueraria edulis)2个物种组成。研究结果显示,锈毛两型豆位于分支1内,其与两型豆亲缘关系最近。
3 讨 论
叶绿体全基因组长度为100~200 kb,包含大量的进化信息,其所含大量的高度可变区域[30],使其成为分子生物标志物、系统发育分析、进化分析和比较基因组研究的理想研究模型[31]。近年来,随着高通量测序技术的高速发展,叶绿体基因组公共数据库相关数据逐步完善,使牧草植物叶绿体基因组测序和研究方面取得了一定的发展,但关于牧草叶绿体基因组的研究仍然不多。目前仅有关于紫花苜蓿(Medicago sativa)、箭筈豌豆(Vicia sativa)、鸭茅(Dactylis glomerata)、三叶草(Trifolium subterraneum)和鹰嘴豆(Cicer arietinum)等少量牧草植物叶绿体基因组的报道研究,牧草在在叶绿体基因组的研究方面还不甚深入,还具有很大的研究发展空间[32]。锈毛两型豆的叶绿体基因组长度为152 531 bp,为典型的双链环状四分体结构,总GC含量为 35.45%,符合常见双子叶植物叶绿体基因组GC总含量在31%~38%的特征[33]。同样的,其反向重复区(41.87%)的总GC含量明显高于大单拷贝区(32.97%)和小单拷贝区(28.81%),其原因可能是因为rRNA基因在该区域的具有较高的 GC值水平表达,而NADH基因可能是导致SSC 区的低GC含量表达的关键[34]。但锈毛两型豆与三叶草、蒺藜苜蓿、鹰嘴豆等传统豆科牧草相比,其叶绿体基因组特征还是具有一定的差异性,这些传统牧草植物因一个反向重复序列( IR)完全丢失而具有特殊的叶绿体基因组结构[32]。
以往的研究表明,简单重复序列(SSRs)在基因组中广泛分布,并且由于其特殊的亲本遗传特征,SSR通常用于遗传群体结构和母体分析,通过寻找叶绿体基因组的SSR,对鉴定植物种质资源和分子标记具有重要意义[35]。本研究结果显示,锈毛两型豆叶绿体基因组共检测到41个单核苷酸、28个二核苷酸SSRs。其中,二核苷酸重复序列共有AT(17 SSRs)和TA(11 SSRs)2种重复类型,表明SSRs 主要由短的 poly A和poly T构成[36]。本研究检测到的简单重复序列可为今后两型豆属植物分子生物学研究提供候选的分子标记。
密码子使用偏好性与不同因素相关,包括基因表达水平、GC含量、氨基酸保护和转录选择等[37]。前人研究表明密码子的使用模式与 GC 含量密切相关,本研究中锈毛两型豆叶绿体基因组48个CDS基因GC1、GC2、GC3和GCall的平均GC含量分别为45.78%(30.8%~56.9%)、38.29%(26.4%~54.7%)、25.67%(18%~ 35.8%)和36.58%(26.8%~43.3%),而两型豆的分别为45.75%(31.1%~56.5%)、38.24%(26%~54.7%)、25.6%(17.5%~35.8%)和36.52%(26.4%~43.3%)。锈毛两型豆和两型豆平均 GC 含量均小于 50%,这说明两种植物的叶绿体基因组倾向于使用A与U作为末尾碱基。这与已报道的与蒺藜苜蓿(Medicago truncatula)[38]和紫苜蓿(Medicago sativa)[39]等表现出一致性。锈毛两型豆和两型豆的RSCU 值相近,锈毛两型豆和两型豆RSCU>1 的密码子均有 11 个,其中有6个密码子以A作为结尾,有5个密码子以U作为结尾,没有以G作为末尾碱基的密码子。而在RSCU<1 的密码子中除UGA和CUA以A结尾外,其余密码子均以 C 或 G 结尾,这与黄花文心兰(Oncidium gower ramsey)[40]、银白杨(Populus alba)[41]和喜马红景天(Rhodiola himalensis)[42]等对叶绿体偏性的分析的结果相似。本研究揭示了选择和突变是影响密码子偏好性的可能因素。选择理论解释说,密码子偏好性有助于蛋白质表达的效率和准确性,因此进行正选择。同时,突变解释假设由于突变模式的非随机性,存在密码子偏倚[43-44]。虽然密码子偏好性选择背后的机制仍有争议,但本研究中确定了GC含量与密码子使用模式之间的强相关性[45]。本研究中锈毛两型豆和两型豆植物叶绿体基因组 RSCU 值的变异范围相似,分别为 0.23~1.92和0.24~1.92,均以CGC(编码精氨酸) 的 RSCU值最低和AGA(编码精氨酸) RSCU值最高,表明锈毛两型豆和两型豆均以CGC使用频率最低,AGA 使用频率最高,这一结果与柿属(Diospyros)[46]植物和蒺藜苜蓿[37]等结果表现出一致性。但却与蒜头果(Malania oleifera)[47]、高山松(Pinus densata)[48]、楸树(Catalpa bungei)[49]等植物的研究表现出差异性,这些植物以AGC(编码丝氨酸)的RSCU值最低,TTA(编码亮氨酸)最高,这表明不同植物的氨基酸与密码子的偏好性具有一定的差异。
根据分子进化的中性理论,碱基突变和自然选择对密码子第3碱基变化的影响是中性的或接近中性的[50]。如果密码子的使用受到自然选择的影响,GC3值的分布范围较小,GC12和GC3没有显著的相关性[51]。本研究表明,锈毛两型豆和两型豆的GC12与 GC3 的相关系数分别为0.341和0.362,相关性均达到了显著水平(P<0.05),说明突变压力对密码子偏好性具有显著的影响。然而,ENC-plot和PR2-plot分析表明,锈毛两型豆和两型豆叶绿体基因组的密码子偏好可能受到突变和自然选择的影响。由此可以看出,植物密码子使用偏差受到多种因素的影响,而影响CUB的主要因素在不同植物物种中也可能存在差异。另外,本研究在锈毛两型豆和两型豆中分别提取到18个和20个最优密码子,其中共有18个共有密码子,两个物种的最优密码子均以A或U结尾,共有最优密码子当中,以A结尾的最优密码子共计7个,以U结尾的有11个。同时本研究在研究锈毛两型豆和两型豆叶绿体基因组的基础上第一次对两型豆属已公布在NCBI平台上的叶绿体基因组序列进行了系统发育分析,确定了两型豆属的系统发育位置。总之,本研究对于两型豆属叶绿体基因组特征、密码子偏好性及系统发育的分析可能对后续的密码子优化、叶绿体基因组改造、探索物种进化和增加外源基因表达具有重要意义。
参考文献 Reference:
[1] KUMAR S,SANE P V. Legumes of South Asia [M]. London:Royal Botanic Gardens,Kew. A Checklist,2003:1-536.
[2] SHAN J G,ZHANG Y,GENG S L,et al. Comparative embryology of aerial and subterranean flowers of Amphicarpaea edgeworthii Benth (Leguminosae) [J]. Acta Scientiarum Naturalium Universitatis Pekinensis,2009,45(3):395-401.
[3] OHASHI H,OHASHI K. A taxonomic revision of Amphicarpaea (Leguminosae) including a pollen morphological comparison with Shuteria [J]. Journal of Japanese Botany,2016,91:231-249.
[4] OHASHI K,OHASHI H. A phylogenetic study of Amphicarpaea with a new genus Afroamphica (Leguminosae Tribe Phaseoleae) [J].Journal of Japanese Botany,2018,93(1):9-17.
[5] 郑月萍. 两型豆(Amphicarpaea edgeworthii Benth.)和野大豆(Glycine soja Sieb. et Zucc.)的生物学特性比较研究[D].浙江金华:浙江师范大学,2012.
ZHENG Y P. Comparative study on biological characteristics of Amphicarpaea edgeworthii Benth. and Glycine sofa Sieb.et Zucc. [D]. Jinhua Zhejiang :Zhejiang Normal University,2012.
[6] 吳征镒.中国植物志(第41卷1分册)[M].北京:科学出版社,1995:257.
WU ZH Y. Flora Reipublicae Popularis Sinicae (Volume 41,Division 1) [M]. Beijing:Science Press,1995:257.
[7] 乔亚科,李桂兰,王文颇,等.三籽两型豆生长发育特性及栽培技术研究[J].中国中药杂志,2003(10):24-26.
QIAO Y K,LI G L,WANG W P,et al. Primary study on developmental speciality and cultural practices of Amphicarpaea edgeworthii[J]. China Journal of Chinese Materia Medica,2003(10):24-26.
[8] 刘国道,杨虎彪.中国南方牧草志.第一卷,豆科[M].北京:科学出版社,2022:479.
LIU G D,YANG H B.Forage Flora of Southern China. Volume 1,Leguminosae [M]. Beijing:Science Press,2022:479.
[9] SUGIURA M,SHINOZAKI K,ZAITA N,et al. Clone bank of the tobacco (Nicotiana tabacum) chloroplast genome as a set of overlapping restriction endonuclease fragments:mapping of eleven ribosomal protein genes [J]. Plant Science,1986,44:211-217.
[10] SUGIURA M. The chloroplast genome [J]. Plant Molecular Biology,1992,19:149-168.
[11] PEDEN J F. Analysis of codon usage [J]. Biology System,1999,5:45-50.
[12] SHARP P M,EMERY L R,ZENG K. Forces that influence the evolution of codon bias [J]. Philosophical Transactions of the Royal Society of London. Series B:Biological Sciences,2010,365:1203-1212.
[13] SHARP P M,STENICO M,PEDEN J F,et al. Codon usage:mutational bias,translational selection,or both? [J]. Biochemical Society Transactions,1993,21:835-841.
[14] SUBRAMANIAN S. Nearly neutrality and the evolution of codon usage bias in Eukaryotic genomes:figure 1 [J]. Genetics,2008,178:2429-2432.
[15] QIN H,WU W B,COMERON J M,et al. Intragenic spatial patterns of codon usage bias in prokaryotic and eukaryotic genomes [J]. Genetics,2004,168:2245-2260.
[16] XING Z B,CAO L,ZHOU M,et al. Analysis on codon usage of chloroplast genome of Eleutherococcus senticosus [J]. China Journal of Chinese Materia Medica,2013, 38(5):661-665.
[17] TAI T H,TANKSLEY S D. A rapid and inexpensive method for isolation of total DNA from dehydrated plant tissue [J]. Plant Molecular Biology Reporter,1990, 8(4):297-303.
[18] DING B C,SUN Y,RONG F X,et al. The complete mitochondrial genome of Holothuria spinifera (Théel,1866) [J].Mitochondrial DNA Part B:Resources,2020,5(2):1679-1680.
[19] QU X J,MOORE M J,LI D Z,et al. PGA:a software package for rapid,accurate,and flexible batch annotation of plastomes [J].Plant Methods,2019,15(1):50.
[20] BEIER S,THIEL T,MUUNCH T,et al. MISA-web:a web server for microsatellite prediction [J]. Bioinformatics,2017,33(16):2583-2585.
[21] ROSENBERG MS,SUBRAMANIAN S,KUMAR S. Patterns of transitional mutation biases within and among mammalian genomes [J]. Molecular Biology and Evolution,2003,20:988-993.
[22] NOBORU S. Directional mutation pressure and neutral molecular evolution [J]. Proceedings of the National Academy of Sciences of the United States of America,1988,85:2653-2657.
[23] XING Y P,XU L,CHEN S Y,et al.Comparative analysis of complete chloroplast genomes sequences of Arctium lappa and A.tomentosum [J]. Biologia Plantarum,2019,63(1):565-574.
[24] WRIGHT F. The ‘effective number of codons used in a gene [J]. Gene,1990,87:23-29
[25] 尚明照,劉 方,华金平,等. 陆地棉叶绿体基因组密码子使用偏性的分析[J].中国农业科学,2011,44(2):245-253.
SHANG M ZH,LIU F,HUA J P,et al. Analysis on codon usage of chloroplast genome of Gossypium hirsutum[J]. Scientia Agricultura Sinica,2011,44(2):245-253.
[26] 赵 洋,刘 振,杨培迪,等.密码子偏性分析方法及茶树中密码子偏性研究进展[J].茶叶通讯,2016,43(2):3-7.
ZHAO Y,LIU ZH,YANG P D,et al. Codon bias analysis method and research progress on codon bias in Camellia sinensis [J]. Journal of Tea Communication,2016, 43(2):3-7.
[27] 胡莎莎,罗 洪,吴 琦,等.苦荞叶绿体基因组密码子偏爱性分析[J].分子植物育种,2016,14(2):309-317.
HU SH SH,LUO H,WU Q,et al. Analysis of codon bias of chloroplast genome of Tartary Buckwheat[J]. Molecular Plant Breeding,2016,14(2):309-317.
[28] KATOH K,MISAWA K,KUMA K I,et al. MAFFT:a novel method for rapid multiple sequence alignment based on fast fourier transform [J]. Nucleic Acids Research,2002,30(14):3059-3066.
[29] NIE X,LV S,ZHANG Y,et al. Complete chloroplast genome sequence of a major invasive species,crofton weed (Ageratina adenophora)[J]. PLoS One,2012,7:e36869
[30] GUO S,LIAO X,CHEN S,et al. A comparative analysis of the chloroplast genomes of four Polygonum medicinal plants[J]. Frontiers in Genetics,2022,13:764534.
[31] TIAN C,LI X,WU Z,et al. Characterization and comparative analysis of complete chloroplast genomes of three species from the Genus Astragalus (Leguminosae)[J]. Frontiers in Genetics,2021,12:705482.
[32] 陶曉丽,王彦荣,刘志鹏.牧草叶绿体基因组研究进展[J].草业科学,2015,32(6):978-987.
TAO X L,WANG Y R,LIU ZH P. Progress in chloroplast genome analysis of herbage [J].Pratacultural Science,2015,32(6):978-987.
[33] 丁祥青,毕远洋,陈佳婷,等.抱茎金花茶(Camellia tienii)的叶绿体基因组特征分析[J].江苏农业科学,2022, 50(23):33-40.
DING X Q,BI Y Y,CHEN J T,et al. Analysis of chloroplast genome characteristics of Camellia tienii[J]. Jiangsu Agricultural Sciences, 2022, 50(23):33-40.
[34] YAN L,WANG H,HUANG X,et al. Chloroplast genomes of Genus Tilia:comparative genomics and molecular evolution[J]. Frontiers in Genetics,2022,13:925726.
[35] LIU S,FENG S,HUANG Y,et al. Characterization of the complete chloroplast genome of Buddleja lindleyana [J]. Journal of AOAC International,2022,105(1):202-210.
[36] KUANG D Y,WU H,WANG Y L,et al. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae):implication for DNA barcoding and population genetics [J]. Genome,2011,54(8):663-673.
[37] 吴宪明,吴松锋,任大明,等. 密码子偏性的分析方法及相关研究进展[J].遗传,2007,29(4):420-426.
WU X M,WU S F,REN D M,et al. The analysis method and progress in the study of codon bias [J]. Hereditas,2007,29(4):420-426.
[38] 杨国锋,苏昆龙,赵怡然,等. 蒺藜苜蓿叶绿体密码子偏好性分析[J].草业学报,2015,24(12):171-179.
YANG G F,SU K L,ZHAO Y R,et al. Analysis of codon usage in the chloroplast genome of Medicago truncatula [J]. Acta Prataculturae Sinica,2015,24(12):171-179.
[39] 喻 凤,韩 明.紫花苜蓿叶绿体基因组密码子偏好性分析[J].广西植物,2021,41(12):2069-2076.
YU F,HAN M. Analysis of codon usage bias in the chloroplast genome of alfalfa (Medicago sativa) [J]. Guihaia,2021,41(12):2069-2076.
[40] XU C,CAI X,CHEN Q. Factors affecting synonymous codon usage bias in chloroplast genome of Oncidium gowerramsey [J]. Evolutionary Bioinformatics,2011,7:271-278.
[41] ZHOU M,LONG W,LI X. Analysis of synonymous codon usage in chloroplast genome of Populas alba [J]. Journal of Forestry Research,2008,4:293-297.
[42] 张 雨,苏 旭,刘玉萍,等.喜马红景天叶绿体基因组特征及其系统发育分析[J].植物研究,2022,42(4):602-612.
ZHANG Y,SU X,LIU Y P,et al. Characteristics of complete chloroplast genome and phylogenetic analysis of Rhodiola himalensis (Crassulaceae) [J]. Bulletin of Botanical Research,2022,42(4):602-612.
[43] LESEBERG C H,DUVALL M R. The complete chloroplast genome ofCoix lacryma-jobi and a comparative molecular evolutionary analysis of plastomes in cereals [J].Journal of Molecular Evolution,2009,69(4):311-318.
[44] BUMER M. The selection-mutation-drift theory of synonymous codon usage[J].Genetics,1991,129:897-907.
[45] GUI L J,JIANG S F,XIE D F,et al. Analysis of complete chloroplast genomes of Curcuma and the contribution to phylogeny and adaptive evolution [J]. Gene,2020,732:144355.
[46] 傅建敏,索玉静,刘慧敏,等.柿属植物叶绿体蛋白质编码基因密码子用法[J].经济林研究,2017,35(2):38-44.
FU J M,SUO Y J,LIU H M,et al. Analysis on codon usage in the chloroplast protein-coding genes of Diospyros spp. [J]. Nonwood Forest Research,2017,35(2):38-44.
[47] 原晓龙,刘 音,康洪梅,等.蒜头果叶绿体基因组密码子偏好性分析[J].西南林业大学学报(自然科学),2021, 41(3):15-22.
YUAN X L,LIU Y,KANG H M,et al. Analysis of codon usage bias in chloroplast genome of Malania oleifera [J]. Journal of Southwest Forestry University,2021,41(3):15-22.
[48] 李江飞,李亚麒,唐军荣,等.高山松叶绿体基因组密码子偏好性模式[J].生物学杂志,2023,40(1):52-59.
LI J F,LI Y Q,TANG J R,et al. Comparison of codon preference patterns in the chloroplast genome of Pinus densata [J]. Journal of Biology,2023,40(1):52-59.
[49] 李 鳳,辛 静,辛雅萱,等. 楸树叶绿体基因组密码子偏性分析[J].南方农业学报,2021,52(10):2735-2743.
LI F,XIN J,XIN Y X,et al. Codon usage bias of chloroplast genome in Catalpa bungei C.A.Mey. [J]. Journal of Southern Agriculture ,2021,52(10):2735-2743.
[50] ZHOU M,LONG W,LI X. Patterns of synonymous codon usage bias in chloroplast genomes of seed plants [J]. For Study China,2008,10(4):235-242.
[51] KAWABE A,MIYASHITA N T.Patterns of codon usage bias in three dicot and four monocot plant species [J]. Genes & Genetic Systems,2003,5:343-352.
Genomic Characteristics and Codon Usage Bias of Chloroplast Genome in Amphicarpaea Elliot
Abstract In order to elucidate the chloroplast genome structure of Amphicarpaea ferruginea and analyze the codon usage bias and influencing factors of Amphicarpaea species chloroplast genome,the leguminous grass species of A. ferruginea which holds considerable potential for development and utilization prospects in the central and southern subtropical regions,was used as the test material. The chloroplast genome of A. ferruginea was sequenced,assembled and annotated using high-throughput sequencing technology,and its chloroplast genome structure and gene composition were analyzed. CodonW 1.4.2 software and CUSP online program were used to analyze the gene codon usage bias parameters and nucleotide composition of A. ferruginea and A. edgeworthii. The results showed that A. ferruginea chloroplast genome had a total length of 152 531 bp and followed a typical tetrad structure,including a large single copy region (LSC) of 83 364 bp,a small single copy region (SSC) of 17 935 bp,and a pair of inverted repeats of 25 616 bp. The GC content was 35.44%. The A. ferruginea chloroplast genome encoded 130 genes, comprising 85 protein-coding genes,37 tRNA genes and 8 rRNA genes. A total of 73 simple sequence repeats (SSRs) were detected in the A. ferruginea chloroplast genome. The numbers of mononucleotide,dinucleotide,trinucleotide,tetranucleotide,pentanucleotide,and hexanucleotide SSRs were 41,28,3,1,0 and 0,respectively. A total of 48 CDS genes suitable for codon usage bias analysis were screened from the A. ferruginea and A. edgeworthii chloroplast genomes. The codon usage patterns of the two plant chloroplast genomes were similar,both tending to use A and U as terminal bases. The ENC values of the 48 genes were all above 35%,indicating that the codon usage bias of A. ferruginea and A. edgeworthii chloroplast genomes was weak. The results of neutral plot analysis,effective number of codons ( ENC-plot ) analysis and parity preference (PR2-plot ) analysis showed that the codon usage patterns of Amphicarpaea chloroplast genome was not only affected by mutation pressure,but also by selection pressure and other factors. 18 and 20 optimal codons were identified in A. ferruginea and A. edgeworthii, respectively,among which there were 18 optimal codons. The phylogenetic tree construction based on the chloroplast genomes of 20 species of Leguminosae showed that the 20 species of Leguminosae constituted a monophyletic group,and A. ferruginea and A. edgeworthii were most closely related.
