铜绿假单胞菌对肺血管内皮细胞功能的影响及其机制
2024-04-10张雷王俊轶何翔吴敏李国平黄锦伟
张雷 王俊轶 何翔 吴敏 李国平 黄锦伟
基金項目:国家自然科学基金(81970026、82000029、82200079)、四川省自然科学基金(2022NSFSC1324)、成都市高水平临床重点专科建设项目(ZX20201202020)和澳门科学技术发展基金(0124/2022/A)
摘要:目的 探究铜绿假单胞菌(PA)感染对肺血管内皮细胞功能的影响,并探讨其加重小鼠肺部炎症反应的可能机制。方法 PAO1菌株感染人肺微血管内皮细胞HULEC-5a 2 h后,采用逆转录实时荧光定量PCR(RT-qPCR)检测自噬相关基因5(ATG5)、6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶3(PFKFB3)和钙黏蛋白5(CDH5)的mRNA表达,免疫荧光检测ATG5、PFKFB3和血管内皮钙黏蛋白(VE-cadherin)的蛋白表达。采用小干扰RNA(siRNA)分别敲减ATG5、PFKFB3后,RT-qPCR检测ATG5、PFKFB3和CDH5的mRNA表达,免疫荧光检测PFKFB3和VE-cadherin的蛋白表达,并采用乳酸测定试剂盒检测细胞内乳酸的水平。采用PAO1菌株感染小鼠后,肺组织切片病理染色观察小鼠肺部炎症,激光共聚焦显微镜观察并分析荧光标记的内皮细胞PFKFB3和VE-cadherin的表达量。结果 与对照组比较,PAO1感染HULEC-5a细胞后,PFKFB3 mRNA和蛋白表达均增加(P均<0.05),CDH5 mRNA表达减少(P=0.023),VE-cadherin蛋白表达减少(P<0.001),且连续性被破坏,乳酸含量升高(P=0.017)。与PAO1感染HULEC-5a细胞比较,PAO1感染敲减PFKFB3的HULEC-5a细胞后,CDH5 mRNA表达增加(P=0.043),VE-cadherin荧光连续性得到恢复,乳酸含量降低(P=0.047);PAO1感染敲减ATG5的HULEC-5a细胞后,PFKFB3 mRNA(P=0.013)和蛋白表达(P=0.003)均增加,CDH5 mRNA(P=0.020)和VE-cadherin蛋白表达(P=0.001)均减少,乳酸含量升高(P=0.015)。PAO1感染小鼠后,病理HE染色结果显示,肺泡内明显的红细胞渗漏,炎症细胞浸润,肺泡间隔增宽和部分血管内皮细胞脱落。免疫荧光染色结果显示,与正常小鼠比较,PAO1感染小鼠内皮细胞PFKFB3表达增加,VE-cadherin荧光减弱,且不连续。结论 PA可能通过AGT5调控PFKFB3通路,破坏肺血管内皮细胞功能,进而加重小鼠肺部炎症反应。
关键词:铜绿假单胞菌;肺血管内皮细胞;6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶3;自噬相关基因5;感染
中图分类号: R378.99+1;R563.1+9 文献标识码: A 文章编号:1000-503X(2024)01-0001-10
DOI:10.3881/j.issn.1000-503X.15831
Pseudomonas Aeruginosa Affects the Function of Pulmonary Vascular Endothelial Cells
ZHANG Lei1,2,3,WANG Junyi1,2,3,HE Xiang2,WU Min3,4,LI Guoping1,2,Vincent Kam Wai Wong1
1State Key Laboratory of Quality Research in Chinese Medicine,Macau University of Science and Technology,Macao 999078,China
2Allergy and Precision Medicine Laboratory,Respiratory Health Research Institute,The Third People‘s Hospital of Chengdu,Chengdu 610000,China
3Department of Biomedical Sciences,School of Medicine and Health Sciences,University of North Dakota,Grand Forks,North Dakota 58203,USA
4Wenzhou Institute of University of Chinese Academy of Sciences,Wenzhou,Zhejiang 325000,China
Corresponding author:Vincent Kam Wai Wong Tel:853-88972408,E-mail:kawwong@must.edu.mo
ABSTRACT:Objective To investigate the impact of Pseudomonas aeruginosa(PA) infection on the function of pulmonary vascular endothelial cells,and explore the mechanism of this bacterium in exacerbating lung inflammation in mice.Methods Two hours after human lung microvascular endothelial cell(HULEC-5a) were infected with the PA strain PAO1,the mRNA levels of autophagy-related gene 5(ATG5),6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3(PFKFB3),and calcium adhesion protein 5(CDH5) were determined by reverse transcription real-time fluorescent quantitative PCR(RT-qPCR).The protein levels of ATG5,PFKFB3,and vascular endothelial calcium adhesion protein(VE-cadherin) were detected by immunofluorescence.After the expression of ATG5 and PFKFB3 was respectively knocked down by small interfering RNA(siRNA),RT-qPCR was employed to measure the mRNA levels of ATG5,PFKFB3,and CDH5,and immunofluorescence to detect the protein levels of PFKFB3 and VE-cadherin.In addition,the lactate assay kit was used to determine the level of lactate in the cells.After mice were infected with PAO1,lung inflammation was assessed through histopathological section staining.Confocal microscopy was employed to capture and analyze fluorescence-labeled PFKFB3 and VE-cadherin in endothelial cells.Results Compared with the control group,the HULEC-5a cells infected with PAO1 showed up-regulated mRNA and protein levels of PFKFB3(all P<0.05),down-regulated mRNA level of CDH5(P=0.023),disrupted continuity and down-regulated protein level of VE-cadherin(P<0.001),and elevated lactate level(P=0.017).Compared with PAO1-infected HULEC-5a cells,knocking down PFKFB3 led to the up-regulated mRNA level of CDH5(P=0.043),lowered lactate level(P=0.047),and restored continuity of VE-cadherin;knocking down ATG5 led to up-regulated mRNA and protein levels of PFKFB3(P=0.013 and P=0.003),elevated lactate level(P=0.015),and down-regulated mRNA level of CDH5(P=0.020) and protein level of VE-cadherin(P=0.001).The HE staining results showed obvious red blood cell leakage,inflammatory cell infiltration,alveolar septal widening,and partial detachment of vascular endothelial cells in the alveoli of PA-infected mice.Immunofluorescence staining showed up-regulated expression of PFKFB3 and decreased fluorescence signal of VE-cadherin in endothelial cells of infected mice compared with normal mice.Conclusion PA may regulate the PFKFB3 pathway via AGT5 to disrupt the function of pulmonary vascular endothelial cells,thereby exacerbating the inflammation in the lungs of mice.
Key words:Pseudomonas aeruginosa;pulmonary vascular endothelial cell;6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3;autophagy-related gene 5;infection
Acta Acad Med Sin,2024,46(1):1-10
铜绿假单胞菌(Pseudomonas aeruginosa,PA)是医院内下呼吸道感染的常见病原菌,在我国医院获得性肺炎病原谱中,PA占16.9%~22.0%,仅次于鲍曼不动杆菌,是重症肺炎、呼吸机相关性肺炎以及结构性肺病患者的重要致病菌[1]。PA致病机制复杂,且存在多种耐药特性,其感染率居高不下。因此,PA是目前院内下呼吸道感染防治的重点和难点[2-3]。探讨PA致病的分子机制和关键调控环节,寻找有效干预靶点,对于院内PA下呼吸道感染的预防和治疗有重要意义。
肺血管内皮细胞是一种半渗透的生物力学屏障,也可以作为一种免疫反应细胞,根据环境条件产生抗炎、促炎和保护反应[4]。内皮细胞能调节血管张力,参与先天免疫及调节细胞与细胞的相互作用和血管壁的细胞代谢等重要生理和病理过程。最近研究认为肺血管内皮细胞是急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)炎症风暴中心[5]。内皮细胞损伤会导致内皮下水肿,加重败血症引起的凝血异常[6]。一些针对内皮功能障碍的药物可以有效治疗心血管疾病和许多其他疾病,包括肺动脉高压、ARDS和肿瘤等[7-8]。据文献报道,PA的膜囊泡可导致内皮屏障损伤和肺衰竭,但具体机制不清[9]。探讨PA感染对肺血管内皮细胞功能障碍的机制,有望为ARDS及脓毒血症等感染性疾病的治疗提供新的视角和方向。6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶3(6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3,PFKFB3)作为糖酵解调节剂,能催化果糖-2,6-二磷酸(F-2,6-P2)的合成,其是糖酵解关键限速酶6-磷酸果糖-1-激酶最有效的变构激活剂[10]。PFKFB3在细胞代谢过程中具有重要的催化作用。在所有组织细胞中,PFKFB3在内皮细胞中表达最显著。已有文献报道PFKFB3介导巨噬细胞的糖酵解,通过影响巨噬细胞的积累和活化在小鼠肺动脉高压的发展中发挥重要作用[11]。然而,关于PFKFB3在感染性疾病中对内皮细胞影响的研究较少。因此,本研究重点探究PA感染后PFKFB3在肺血管内皮细胞功能障碍中的作用及可能机制,为PA相关疾病的治疗提供新的靶点。
1 材料和方法
1.1 材料
PA01菌株由美國马萨诸塞州波士顿哈佛医学院Lory S博士提供,北达科他大学医学院Wu M博士实验室保留。细菌在溶原性肉汤中生长,在37 ℃下振荡培养至光密度值600在0.6~0.8之间。人肺微血管内皮细胞HULEC-5a购自美国ATCC细胞库,逆转录试剂盒购自日本Takara公司,逆转录实时荧光定量PCR(reverse transcription quantitative real-time PCR,RT-qPCR)试剂盒购自美国Applied Biosystems公司,L-乳酸检测试剂盒(货号:ab65330)购自美国Abcam公司,PFKFB3购自美国Proteintech公司,自噬相关基因5(autophagy-related gene 5,ATG5)、血管内皮钙黏蛋白(vascular endothelial cadherin,VE-cadherin)购自美国Cell Signaling公司,TRIzol试剂、Lipofectamine RNAiMAX转染试剂、山羊抗兔Alexa FluorTM 488和594抗体购自美国Invitrogen公司,DAPI购自美国Thermo Fisher公司。激光共聚焦显微镜购自日本Olympus公司,CFX ConnectTM荧光定量PCR检测系统购自美国Bio-Rad公司。
1.2 实验动物及分组
6~8周龄C57BL/6J雄性小鼠购自美国Jackson实验室,饲养于美国北达科他大学实验动物中心SPF级动物房。本研究经北达科他大学动物管理和使用委员会批准,并根据动物护理和机构指南进行规范操作。10只C57BL/6J小鼠随机等分为PAO1处理的感染组(PA感染组)和PBS处理的对照组,适应性喂养1周后,用氯胺酮(80 mg/kg)和甲苯噻嗪(10 mg/kg)麻醉小鼠,PA组采用PBS稀释的30 μL 3×107 CFU的PAO1经气道滴注感染小鼠,对照组用等体积的PBS做相同处理,24 h后,小鼠经CO2麻醉脱颈处死,留取肺组织做后续实验。
1.3 细胞培养及处理
HULEC-5a细胞使用含10%胎牛血清、10 ng/mL表皮生长因子、1 μg/mL氢化可的松、10 mmol/L L-谷氨酰胺、1%链霉素和青霉素的MCDB131培养基,置于5% CO2、37 ℃恒温恒湿细胞培养箱中培养。以2.5×105个/孔的密度均匀接种于6孔板中,分别用感染复数(multiplicity of infection,MOI)为10和20的PAO1感染HULEC-5a细胞2、4 h后,收集细胞进行后续实验。
1.4 细胞转染
HULEC-5a细胞以2.5×105个/孔的密度均匀接种于6孔板中,培养12 h后,按照RNAiMAX转染试剂商品说明书,用每孔30 pmol的小干扰RNA(small interfering RNA,siRNA)转染细胞,6~8 h后更换培养基进行后续实验。si-ATG5和si-PFKFB3均由美国Santa Cruz生物公司合成。
1.5 RT-qPCR实验
采用TRIzol试剂提取细胞总RNA,按照TAKARA逆转录试剂盒说明书合成cDNA,并以cDNA为模板进行RT-qPCR。反应条件:95 ℃变性30 s,95 ℃变性5 s,60 ℃退火34 s,共40个循环。以β-actin为内参基因,采用2-ΔΔCt法计算PFKFB3、ATG5、CDH5的相对表达量。引物序列:ATG5上游引物5-GGACGAAACAGC-TTCTGAAT-3,下游引物5-GATGGGATTGCAAAATGACA-3;PFKFB3上游引物5-CCATGAAAGTCCGGAAGCAATG-3,下游引物5-GCTTTTGACATCTCTCAAGGCAG-3;CDH5上游引物5-TTGGAACCAGA-TGCACATTGAT-3,下游引物5-TCTTGCGACTCACG-CTTGAC-3;β-actin上游引物5-AAATCTGGCACCAC-ACCTTC-3,下游引物5-GGGGTGTTGAAGGTC-TCAAA-3。
1.6 细胞免疫荧光染色
HULEC-5a细胞以1.0×105个/孔的密度均匀接种于24孔板中,PAO1感染处理后,4%多聚甲醛固定15 min,0.05% Tween 20透膜10 min,10%山羊血清室温下封闭1 h,然后分别加入VE-cadherin抗体(1∶300)、PFKFB3抗体(1∶200)、ATG5(1∶200)4 ℃孵育过夜,加入二抗室温孵育2 h,DAPI对细胞核进行染色,抗荧光淬灭剂封片后,采用激光共聚焦显微镜观察,拍照记录。
1.7 乳酸含量检测(荧光标定法)
在10 cm的细胞培养皿上接种1.0×106个HULEC-5a细胞,PAO1感染处理后,裂解细胞,4 ℃离心,用4 mol/L高氯酸和2 mol/L氢氧化钾对上清液进行脱蛋白,按照L-乳酸检测试剂盒(荧光标定法)说明书在96孔板上样,在激发波长535 nm、吸收波长587 nm处检测光密度值。每个实验独立重复3次,分析细胞内乳酸的含量。
1.8 病理染色
小鼠处死后取部分右肺,4%多聚甲醛溶液固定48 h后,70%乙醇脱水,石蜡包埋,制备成4 μm厚度的切片,行HE染色,光学显微镜下观察支气管及血管周围肺组织炎症细胞浸润,拍照记录。根据支气管周围炎症的严重程度进行评分:0分:正常;1分:仅有少量炎症细胞;2分:1圈1个细胞层深的炎症细胞;3分:1圈2~4个细胞层深的炎症细胞;4分:1圈>4个细胞层深的炎症细胞[12]。
1.9 肺组织多重免疫荧光染色
小鼠肺组织冰冻切片室温复温后,0.05% Tween 20透膜10 min,10%山羊血清室温孵育1 h封闭非特异性抗体,然后分别加入VE-cadherin抗体(1∶400)、PFKFB3抗体(1∶200)4 ℃孵育过夜,加入二抗室温孵育2 h,DAPI染色,抗荧光淬灭剂封片后,置于激光共聚焦显微镜下观察。
1.10 统计学处理
采用Graphpad Prism 8.0.2软件,对所有数据进行DAgostino-Pearson正态性检验,符合正态分布且方差齐的计量资料以均数±标准误表示,两独立样本比较采用t检验,多组间比较采用ANOVA单因素方差分析和Tukey-Kramer检验。双侧P<0.05为差异有统计学意义。
2 结果
2.1 PAO1感染对HULEC-5a细胞PFKFB3表达量的影响
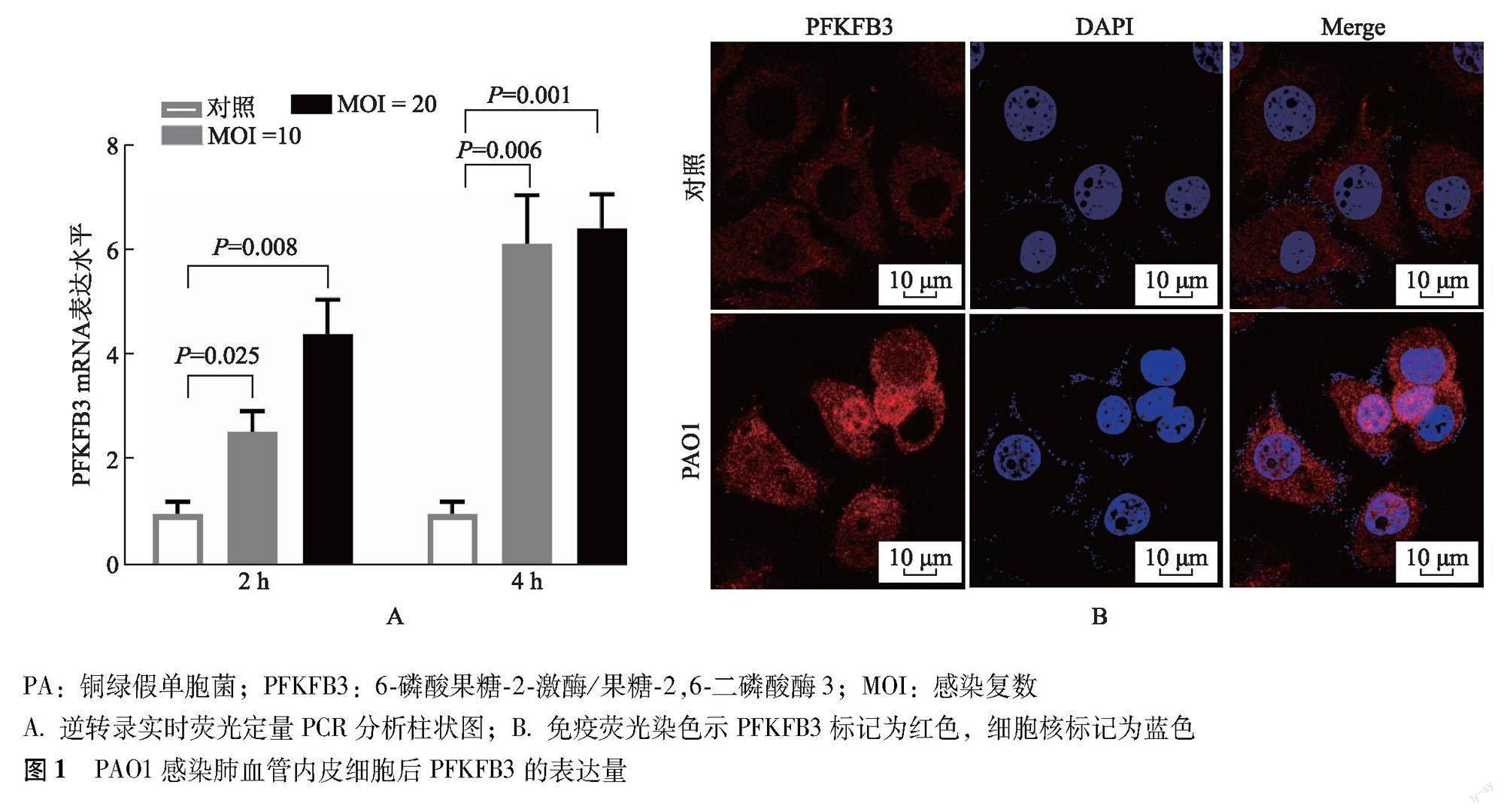
RT-qPCR结果显示,与对照组比较,MOI值为10、20的PAO1处理2 h后,HULEC-5a细胞PFKFB3 mRNA表达显著增加(P=0.025,P=0.008),处理4 h后PFKFB3 mRNA表达显著增加(P=0.006,P=0.001)(图1A)。采用PAO1(MOI=10)感染HULEC-5a细胞2 h后,免疫荧光染色结果显示,PAO1组细胞PFKFB3蛋白荧光表达较对照组显著增加(701.88±109.41比471.57±105.16,P<0.001)(图1B)。
2.2 PAO1感染HULEC-5a细胞中PFKFB3对VE-cadherin表达的影响
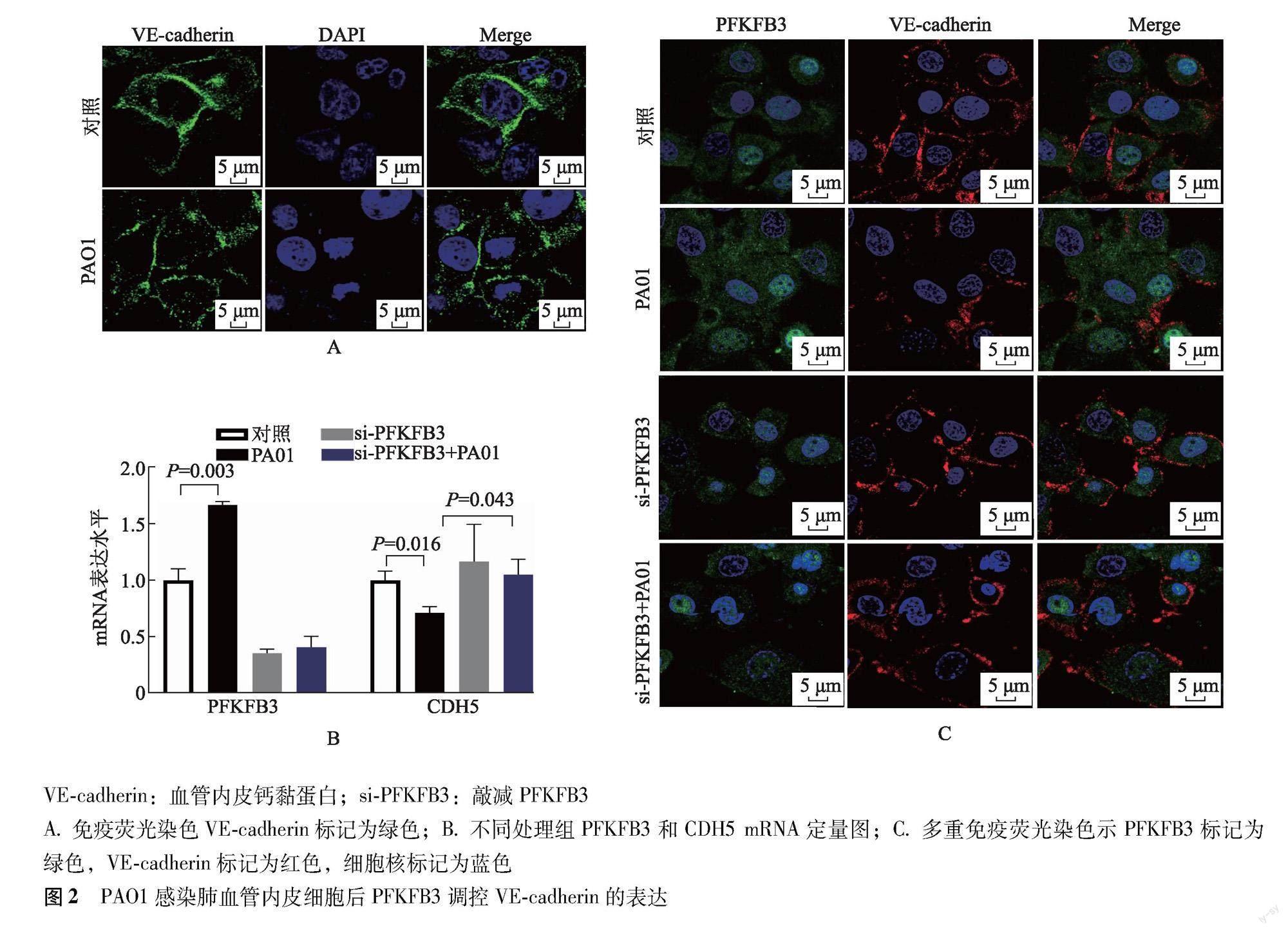
免疫荧光染色结果显示,PAO1组细胞VE-cadherin荧光表达显著低于对照组(333.33±50.67比584.45±58.62,P<0.001),且荧光连续性被破坏(图2A)。RT-qPCR结果显示,与对照组比较,PAO1组细胞CDH5 mRNA表达显著减少(0.62±0.10比1.00±0.10,P=0.016);与PAO1组比较,si-PFKFB3+PAO1组细胞CDH5 mRNA表达增加(1.05±0.14比0.71±0.06,P=0.043)(图2B)。多重免疫荧光染色结果显示,si-PFKFB3+PAO1组细胞VE-cadherin熒光表达显著高于PA01组(445.23±79.76比333.33±50.67,P<0.001)(图2C),且荧光连续性部分恢复。
2.3 PAO1感染HULEC-5a细胞中ATG5对PFKFB3表达的影响
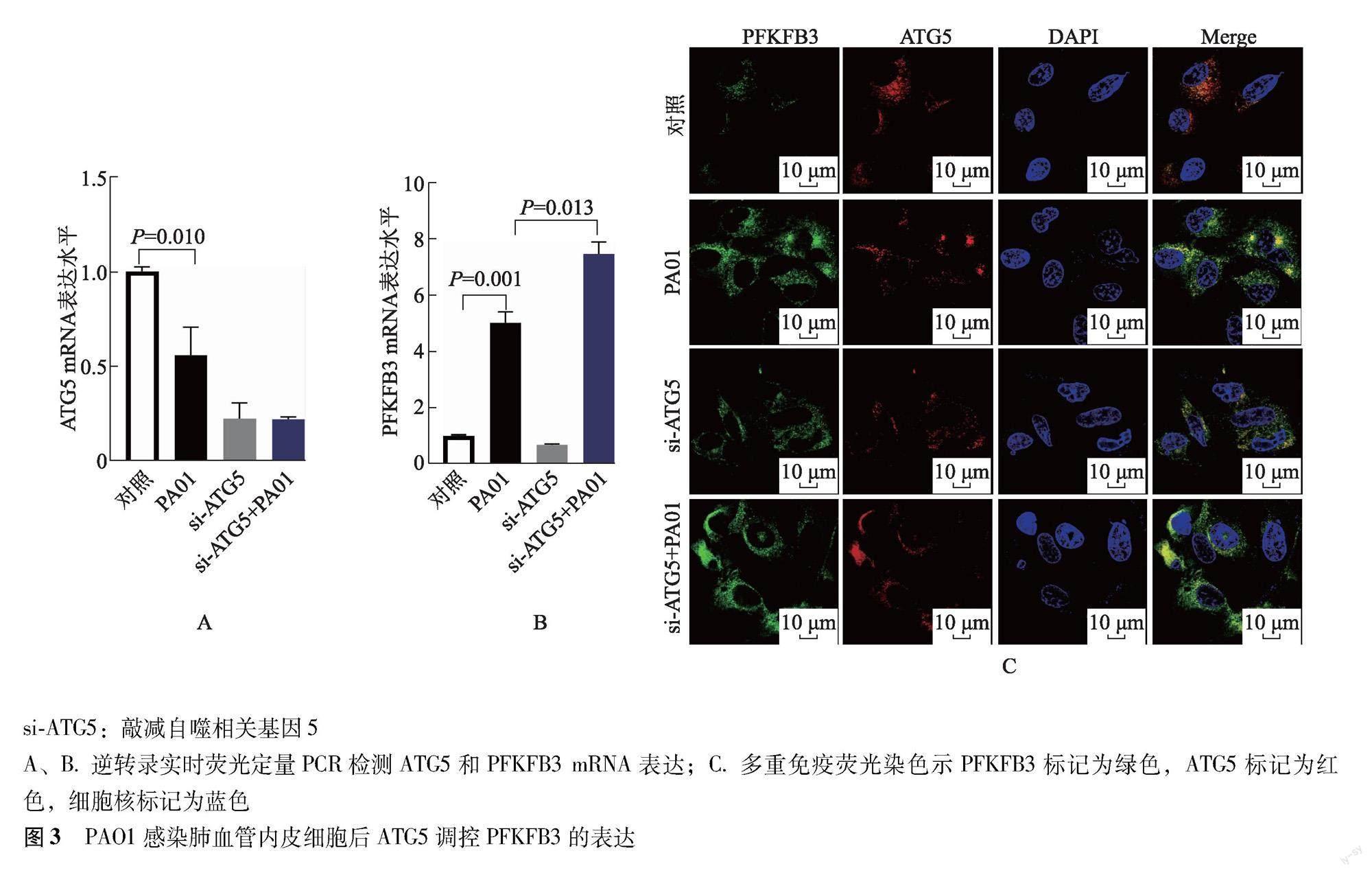
RT-qPCR结果显示,与对照组比较,PAO1组细胞ATG5 mRNA表达减少(0.78±0.07比1.00±0.02,P=0.010)(图3A);与PAO1组比较,si-PFKFB3+PAO1组细胞PFKFB3 mRNA表达增加(7.46±0.43比5.01±0.39,P=0.013)(图3B)。多重免疫荧光染色结果显示,与对照组比较,PAO1组细胞PFKFB3绿色荧光表达增加,ATG5红色荧光表达减少;si-PFKFB3+PAO1组细胞PFKFB3蛋白荧光表达显著高于PAO1组(837.26±62.12比730.97±80.55,P=0.003)(图3C)。
2.4 PAO1感染HULEC-5a细胞中ATG5介导PFKFB3对细胞乳酸代谢的影响
与对照组比较,PAO1组细胞内乳酸水平升高(2.70±0.41比1.72±0.11,P=0.017);与PAO1组比较,si-PFKFB3+PAO1组细胞乳酸水平降低(2.01±0.07比2.70±0.41,P=0.047),si-ATG5+PAO1组细胞乳酸水平升高(3.06±0.03比2.22±0.35,P=0.015)。
2.5 PAO1感染HULEC-5a细胞中ATG5对VE-cadherin表达的影响
RT-qPCR结果显示,si-ATG5+PAO1组细胞CDH5 mRNA表达较PAO1组减少(0.60±0.02比0.77±0.06,P=0.020)(图4A)。多重免疫荧光染色结果显示,si-ATG5+PAO1组细胞VE-cadherin蛋白红色荧光表达显著低于PAO1组(489.12±94.93比592.31±36.70,P=0.001)(图4B、4C)。
2.6 PAO1感染小鼠对肺血管内皮细胞自噬和PFKFB3表达的影响
HE染色结果显示,PA感染组小鼠肺组织可见大量炎性细胞浸润,肺泡间隔增宽,肺泡壁结构破坏,肺泡壁及肺泡腔可见较多红细胞,而对照组未见上述异常改变(图5A)。炎症评分结果显示,与对照组比较,PA感染组炎症评分明显增高(3.85±0.25比0.75±0.19,P<0.001)(图5B)。多重免疫荧光染色显示,PA感染组小鼠肺血管内皮细胞VE-cadherin荧光较对照组减弱,且不连续;PFKFB3荧光较对照组明显增强(图5C)。
3 讨论
PA是一种革兰氏阴性机会致病菌,由于处于生物膜状态的PA能够在低氧或其他恶劣的环境中存活,包括在潮湿的表面上繁殖,所以其可以广泛存在于医疗设备中[13],患有慢性阻塞性肺疾病、囊性纤维化、癌症、烧伤、脓毒血症和呼吸机相关性肺炎等免疫功能低下的患者容易发生急性或慢性PA感染。PA通过分泌多种毒力因子来适应宿主体内的不利环境,从而使宿主成功感染并致病,如脂多糖、外膜蛋白、鞭毛、菌毛和其他黏附素、6型分泌系统等。宿主的先天免疫系统通过多种机制来抵御细菌的感染,包括模式识别受体、质膜信号、细胞内酶和细胞因子/趋化因子参与的炎症反应。同时,生物信息学、代谢组学、单细胞测序、纳米颗粒、药物筛选和噬菌体治疗等新兴技术,也已被用于对PA发病机制和宿主防御的研究中。然而,PA致病与宿主免疫应答之间的关系仍有待探究[14-15]。2023年中国细菌耐药监测网(http://www.chinets.com/)数据显示,从临床分离的致病菌株中,PA是继大肠杆菌、肺炎克雷伯菌、金黄色葡萄球菌和鲍曼不动杆菌之后的第5种医院感染病原体,占7.46%。截至2023年,对碳青霉烯类耐药的PA检出率为23.3%,对替卡西林/克拉维酸耐药的PA检出率为30.8%。因此,耐药PA感染的治疗面临巨大挑战[16-17]。探索PA的致病机制,确定PA潜在的药物治疗靶点,开发新的抗生素和有效疫苗,是当下科研人员和临床医生关注的焦点和亟待解决的科学问题。
内皮细胞是血管壁上的单层细胞,通过动态调节血管张力、血管生成、止血以及抗氧化、抗炎和抗血栓等,在维持多器官正常生理功能和稳态方面发挥着关键作用。血管内皮功能障碍表现为内皮依赖性血管舒张受损、慢性炎症、白细胞黏附和过度渗透以及细胞衰老等。与内皮细胞的其他功能相比,内皮细胞代谢是一个相对较新的研究领域[18]。内皮细胞的主要能量来源是糖酵解。研究发现人脐静脉内皮细胞中约80%的三磷酸腺苷是通过糖酵解途径产生的[19]。在糖酵解过程中,85%的ATP是在PFKFB3催化下通过将葡萄糖转化为乳酸产生的[20]。PFKFB3缺陷的内皮细胞中磷酸化丝氨酸/苏氨酸激酶水平较低,细胞内乳酸显著降低[21]。血清中乳酸水平被认为是组织缺氧的标志物[22]。本研究结果发现,PA感染后PFKFB3调控的糖酵解代谢增加。
内皮细胞的代谢重编程和免疫防御功能存在关联[23-24]。研究表明PFKFB3不僅调控感染后内皮细胞的代谢活动,
也可能参与内皮细胞抗炎及黏附等功能[25]。内皮细胞PFKFB3通过活化B细胞的核因子-κ轻链增强子信号通路调控黏附因子的表达及炎症因子的释放,从而促进由于过度炎症反应造成的组织细胞损伤,PFKFB3敲减可减轻小鼠内毒素诱导的急性肺损伤。内皮细胞间连接由黏附、紧密和间隙连接的蛋白质复合物组成[26]。VE-cadherin完整性受损导致黏附连接分解,是造成病理状态下组织水肿的主要原因。在败血症、局部缺血和创伤等疾病中,内皮细胞屏障完整性的破坏均会导致血管高渗透性和组织肿胀/水肿[27]。本研究中,PA感染通过PFKFB3途径影响内皮细胞的代谢,促进内皮细胞黏附连接受损,加重组织水肿和炎症反应。
自噬作为一种溶酶体依赖性分解代谢过程,被认为是维持能量稳态的微调和关键过程[28]。自噬维持线粒体代谢以满足生物合成和能量需求[29]。研究显示大鼠肉瘤激活的肺癌细胞敲减ATG5和ATG7后会损害线粒体功能,导致线粒体膜电位丧失和线粒体呼吸、细胞能量以及三羧酸循环代谢物的水平降低[30]。自噬损伤可参与内皮-间质转化的细胞代谢紊乱[13]。巨噬细胞可以通过糖酵解重编程以响应自噬、炎性因子激活和细胞焦亡来清除细菌[31]。研究发现,PFKFB3能够与自噬复合体中螯合体1蛋白的泛素结合域进行结合,降低PFKFB3的表达,并诱发乳腺癌干细胞进入休眠状态[32]。同时,也有研究发现,PFKFB3通过调节自噬,影响缺氧诱导的肺动脉高压中的血管重塑[33]。本研究中,PA感染后内皮细胞可以通过自噬关键基因ATG5调控PFKFB3介导的代谢重编程,进而调节内皮细胞黏附连接,维持内皮细胞的屏障功能。因此,PA感染后内皮细胞通过自噬抑制PFKFB3表达是一种自我保护的调控机制。据文献报道,平滑肌细胞特异性敲减PFKFB3后,能够抑制肺动脉高压发展过程中的肺血管重构[11,25,33]。其机制可能是通过降低生长因子、促炎细胞
因子和細胞黏附因子的表达,抑制缺氧诱导的肺动脉高压的进展,从而减轻内毒素诱导的小鼠急性肺损伤。
综上,本研究结果表明,PA感染后肺血管内皮细胞通过ATG5调控PFKFB3表达,促进细胞无氧糖酵解,导致细胞代谢紊乱,内皮细胞间连接被破坏,导致小鼠肺组织水肿,炎症反应加重。PFKFB3有可能成为一个治疗PA下呼吸道感染的潜在靶点。
利益冲突 所有作者声明无利益冲突
作者贡献声明 张雷、王俊轶:设计并实施实验、统计数据、撰写论文;何翔、吴敏、李国平:设计实验、指导研究、提供基金支持;黄锦伟:指导研究、修改论文
参 考 文 献
[1]中华医学会呼吸病学分会感染学组.中国成人医院获得性肺炎与呼吸机相关性肺炎诊断和治疗指南(2018年版)[J].中华结核和呼吸杂志,2018,41(4):255-280.DOI:10.3760/cma.j.issn.1001-0939.2018.04.006.
[2]Daikos GL,da Cunha CA,Rossolini GM,et al.Review of ceftazidime-avibactam for the treatment of infections caused by Pseudomonas aeruginosa[J].Antibiotics,2021,10(9):1126.DOI:10.3390/antibiotics10091126.
[3]Botelho J,Grosso F,Peixe L.Antibiotic resistance in Pseudomonas aeruginosa-mechanisms,epidemiology,and evolution[J].Drug Resist Updat,2019,44:100640.DOI:10.1016/j.drup.2019.07.002.
[4]Xu S,Ilyas I,Little PJ,et al.Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond:from mechanism to pharmacotherapies[J].Pharmacol Rev,2021,73(3):924-967.DOI:10.1124/pharmrev.120.000096.
[5]Suzuki K,Okada H,Takemura G,et al.Recombinant thrombomodulin protects against LPS-induced acute respiratory distress syndrome via preservation of pulmonary endothelial glycocalyx[J].Br J Pharmacol,2020,177(17):4021-4033.DOI:10.1111/bph.15153.
[6]Luo X,Cai S,Li Y,et al.Drp-1 as potential therapeutic target for lipopolysaccharide-induced vascular hyperpermeability[J].Oxid Med Cell Longev,2020,2020:5820245.DOI:10.1155/2020/5820245.
[7]Joshi AD,Dimitropoulou C,Thangjam G,et al.Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling[J].Am J Respir Cell Mol Biol,2014,50(1):170-179.DOI:10.1165/rcmb.2012-0496OC.
[8]Incalza MA,DOria R,Natalicchio A, et al.Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases[J].Vascul Pharmacol,2018,100:1-19.DOI:10.1016/j.vph.2017.05.005.
[9]McVey MJ,Maishan M,Foley A,et al.Pseudomonas aeruginosa membrane vesicles cause endothelial barrier failure and lung injury[J].Eur Respir J,2022,59(5):2101500.DOI:10.1183/13993003.01500-2021.
[10]Jiang H,Shi H,Sun M,et al.FKFB3-Driven macrophage glycolytic metabolism is a crucial component of innate antiviral defense[J].J Immunol,2016,197(7):2880-2890.DOI:10.4049/jimmunol.1600474.
[11]Cao Y,Zhang X,Wang L,et al.PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension[J].Proc Natl Acad Sci USA,2019,116(27):13394-13403.DOI:10.1073/pnas.1821401116.
[12]Kang JY,Lee SY,Rhee CK,et al.Effect of aging on airway remodeling and muscarinic receptors in a murine acute asthma model[J].Clin Interv Aging,2013,8:1393-1403.DOI:10.2147/CIA.S50496.
[13]Sinha M,Ghosh N,Wijesinghe DS,et al.Pseudomonas Aeruginosa theft biofilm require host lipids of cutaneous wound[J].Ann Surg,2023,277(3):e634-e647.DOI:10.1097/SLA.0000000000005252.
[14]Jurado-Martín I,Sainz-Mejías M,McClean S.Pseudomonas aeruginosa:an audacious pathogen with an adaptable arsenal of virulence factors[J].Int J Mol Sci,2021,22(6):3128.DOI:10.3390/ijms22063128.
[15]Qin S,Xiao W,Zhou C,et al.Pseudomonas aeruginosa:pathogenesis,virulence factors,antibiotic resistance,interaction with host,technology advances and emerging therapeutics[J].Signal Transduct Target Ther,2022,7(1):199.DOI:10.1038/s41392-022-01056-1.
[16]Jangra V,Sharma N,Chhillar AK.Therapeutic approaches for combating Pseudomonas aeruginosa infections[J].Microbes Infect,2022,24(4):104950.DOI:10.1016/j.micinf.2022.104950.
[17]Blomquist KC,Nix DE.A critical evaluation of newer β-lactam antibiotics for treatment of Pseudomonas aeruginosa infections[J].Ann Pharmacother,2021,55(8):1010-1024.DOI:10.1177/1060028020974003.
[18]Marzoog BA.Recent advances in molecular biology of metabolic syndrome pathophysiology:endothelial dysfunction as a potential therapeutic target[J].J Diabetes Metab Disord,2022,21(2):1903-1911.DOI:10.1007/s40200-022-01088-y.
[19]Bierhansl L,Conradi LC,Treps L,et al.Central role of metabolism in endothelial cell function and vascular disease[J].Physiology(Bethesda),2017,32(2):126-140.DOI:10.1152/physiol.00031.2016.
[20]De Bock K,Georgiadou M,Schoors S,et al.Role of PFKFB3-driven glycolysis in vessel sprouting[J].Cell,2013,154(3):651-663.DOI:10.1016/j.cell.2013.06.037.
[21]Xu Y,An X,Guo X,et al.Endothelial PFKFB3 plays a critical role in angiogenesis[J].Arterioscler Thromb Vasc Biol,2014,34(6):1231-1239.DOI:10.1161/ATVBAHA.113. 303041.
[22]Nolt B,Tu F,Wang X,et al.Lactate and immunosuppression in sepsis[J].Shock,2018,49(2):120-125.DOI:10.1097/SHK.0000000000000958.
[23]Codo AC,Davanzo GG,Monteiro LB,et al.Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis[J].Cell Metab,2020,32(3):437-446.e5.DOI:10.1016/j.cmet.2020.07.007.
[24]Xiao W,Oldham WM,Priolo C,et al.Immuno-metabolic endothelial phenotypes:integrating inflammation and glucose metabolism[J].Circ Res,2021,129(1):9-29.DOI:10.1161/CIRCRESAHA.120.318805.
[25]Wang L,Cao Y,Gorshkov B,et al.Ablation of endothelial Pfkfb3 protects mice from acute lung injury in LPS-induced endotoxemia[J].Pharmacol Res,2019,146:104292.DOI:10.1016/j.phrs.2019.104292.
[26]Pappenheimer JR,Renkin EM,Borrero LM.Filtration,diffusion and molecular sieving through peripheral capillary membranes;a contribution to the pore theory of capillary permeability[J].Am J Physiol,1951,167(1):13-46.DOI:10.1152/ajplegacy.1951.167.1.13.
[27]Komarova YA,Kruse K,Mehta D,et al.Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability[J].Circ Res,2017,120(1):179-206.DOI:10.1161/CIRCRESAHA.116.306534.
[28]Klionsky DJ,Emr SD.Autophagy as a regulated pathway of cellular degradation[J].Science,2000,290(5497):1717-1721.DOI:10.1126/science.290.5497.1717.
[29]Reyes-Castellanos G,Abdel Hadi N,Carrier A.Autophagy contributes to metabolic reprogramming and therapeutic resistance in pancreatic tumors[J].Cells,2022,11(3):426.DOI:10.3390/cells11030426.
[30]Guo JY,Teng X,Laddha SV,et al.Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells[J].Genes Dev,2016,30(15):1704-1717.DOI:10.1101/gad.283416.116.
[31]Ma L,Li W,Zhang Y,et al.FLT4/VEGFR3 activates AMPK to coordinate glycometabolic reprogramming with autophagy and inflammasome activation for bacterial elimination[J].Autophagy,2022,18(6):1385-1400.DOI:10.1080/15548627.2021.1985338.
[32]La Belle Flynn A,Calhoun BC,Sharma A,et al.Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression[J].Nat Commun,2019,10(1):3668.DOI:10.1038/s41467-019-11640-9.
[33]Kovacs L,Cao Y,Han W,et al.PFKFB3 in smooth muscle promotes vascular remodeling in pulmonary arterial hypertension[J].Am J Respir Crit Care Med,2019,200(5):617-627.DOI:10.1164/rccm.201812-2290OC.
(收稿日期:2023-09-06)
勘 誤
2022年第44卷第5期794~801页《异丹叶大黄素对脂多糖诱导的小鼠急性肺损伤的影响》一文,图7更改如下:
