基于牡丹涝害胁迫的转录组分析及SSR引物开发
2024-04-08刘慧春许雯婷周江华张加强史小华朱开元
刘慧春,许雯婷,周江华,张加强,史小华,朱开元
(浙江省园林植物与花卉研究所,浙江 杭州 311251)
牡丹(PaeoniasuffruticosaL.)是原产于中国的著名花灌木,也是一种具有深厚文化内涵的中国传统花卉。牡丹原产于北方,主要种植于黄河流域中下游地区,根系肥厚,对涝害较为敏感。长江以南地处长江中下游,地下水位偏高,雨季土壤水分过饱和,台风、洪水等灾害严重影响了长江以南牡丹的种植和应用。随着人们欣赏水平的提高,迫切需要筛选出适合江南地区广泛种植的牡丹品种。耐涝品种的选育一直是牡丹南下栽培的重点和难点。目前,江南牡丹的种植主要采用塑料薄膜防雨,限制了植物的自然光照,从而影响了牡丹的开花质量。如何提高牡丹的耐涝性已成为牡丹生产中亟待解决的问题。因此,深入研究牡丹在涝害胁迫下的响应机制,对牡丹生产具有重要的现实意义和应用价值。
在过去的几年里,转录组测序技术已被广泛应用于功能基因组研究。随着新一代测序的快速发展,RNA-Seq已成为一种有效的转录组数据分析方法。此外,它还被用来阐明对各种非生物胁迫的反应机制,包括干旱、低温、盐碱和涝害[1-7]。到目前为止,很少有关于牡丹涝害胁迫相关分子机制的报道。仅见报道的有,通过全长转录组测序和第二代转录组测序的结合,初步探讨了凤丹牡丹对淹水胁迫反应的分子机制[8]。因此,对牡丹响应涝害胁迫的综合转录组学分析还有待进一步研究和提升。
随着高通量测序技术的快速发展,进一步延伸出利用转录组序列开发SSR标记,并已被广泛应用于群体遗传学、遗传连锁图的构建,种质与标记辅助选择育种之间的遗传关系分析[9-12]。前人也曾通过RNA-Seq开发出牡丹的一些SSR标记[13]。例如,前人在滇牡丹(P.delavayi)中开发了10个SSR标记[14],在紫斑牡丹(P.rockii)中开发了11个与农艺性状相关的SSR标记、44个与种子性状相关的SSR标记[15-16],在牡丹(P.suffruticosa)中开发了12个与花色形成有关的SSR标记[17]。然而,与玉米(Zeamays)和大豆(Glycinemax)等作物相比,牡丹中目前的SSR标记数量仍然有限,而且关于牡丹涝害胁迫相关基因的SSR分子标记的报道很少。
在本研究中,通过RNA-Seq对耐涝牡丹根的转录组进行了分析,以阐明其对涝害胁迫反应的分子机制。基于构建的高通量RNA-Seq数据库,在牡丹中开发了调控涝害胁迫性状的SSR标记。该研究结果对牡丹进行了大规模转录组注释,将为未来耐涝牡丹品种的育种研究提供宝贵的基因组资源,并为进一步研究其遗传多样性、功能基因的发现和定位、种质资源鉴定和标记辅助选择提供科学依据。
1 材料与方法
1.1 植物材料及处理
实验材料为3年生凤丹牡丹的实生苗,由浙江省园林植物与花卉研究所提供。选择50株在人工涝害胁迫处理下能够存活下来的植株,在含有泥炭、珍珠岩和园土(体积比2∶1∶1)的培养基质中培养。培养条件为:光照14 h,温度25 ℃,光照强度为160 μmol·m-2·s-1光子,相对湿度为50%~60%。将其中25株幼苗作为处理组,放置在土壤表面上方含有3 cm水深的泡沫箱中。将另外25株未经处理的幼苗置于正常培养条件下作为对照。处理48 h后,从两个处理(对照和处理的植株)中采集幼根,然后在液氮中快速冷冻并在-80 ℃下储存备用。
1.2 RNA提取
将对照样品(CK1、CK2和CK3)和涝害处理的样品(W1、W2、W3)的根作为3个生物学重复。根据产品说明书,使用植物总RNA纯化试剂盒(GeneMark TR02)分离总RNA。通过变性琼脂糖凝胶电泳检测RNA质量,并使用NanoDrop 2000分光光度计(Thermo Scientific,USA)测定RNA浓度。
1.3 RNA序列文库的构建
从经过正常条件(对照样品CK1、CK2、CK3)和涝害条件(涝害处理的样品W1、W2、W3)的牡丹根中分离的6个高质量RNA样品,分别用于cDNA文库的构建,然后将文库加入高通量测序平台(Illumina HiSeq 2500,USA)进行测序。cDNA文库的构建方法和Illumina深度测序的过程参考Li等[18]的方法。RNA-Seq读数通过Illumina数据处理软件(Version 1.8)生成。
1.4 数据组装、读取和基因注释
按照Li等[18]描述的方法进行转录组数据组装,采用NR、Swissport、KOG和KEGG数据库对基因功能进行注释,这些数据库通过BLASTA进行比对,截断E值≤1.0-5。
1.5 差异表达基因分析
基于上述获得的RNA-Seq数据,采用在线工具degust (http://vicbioinformatics.com/degust/) 生成主成分分析(PCA)图。通过软件包RPKM评估和量化基因表达水平,并使用Cufflinks软件的Cuffdiff程序(v2.0.2,http://cole-trapnell-lab.github.io/cufflinks/),使用FDR调整基因的多次测试和分析中的P阈值。FDR调整后的P≤0.05和log2R倍数变化的绝对值≥1(倍数变化≥2)被设定为显著差异表达的阈值。基于Wallenius非中心超基因组分布的GOseq R软件包对DEG进行GO富集分析,该软件包可以调整DEG中的基因长度偏差。采用KOBAS软件测试KEGG途径中DEG的统计富集。
1.6 qRT-PCR分析
随机选择14个来自所有DEG的单基因,使用qPCR验证基于RNA-Seq的表达水平分析的可靠性和准确性。利用Oligo 7.57软件设计单基因的引物,按照Li等[18]描述的方法进行qRT-PCR验证。PCR反应体系为20 μL,包括:1 μL cDNA模板、1 μL引物混合物、10 μL 2×SYBR Green qPCR混合物和8 μL ddH2O。用Mastercycler®ep realplex4进行qPCR反应,反应程序如下:95 ℃预变性3 min;95 ℃变性5 s,60 ℃退火10 s,40个循环;最后95 ℃延伸15 s。选择18S rRNA作为参考基因,并使用2-ΔΔCT方法对基因的相对表达水平进行测算。
1.7 SSR鉴定、引物设计和PCR扩增
根据RNA-Seq数据,使用SSRIT软件(http://archive/gramene.org/db/markers/ssrtool)鉴定SSR位点[17]。已鉴定的基因位点包含二、三、四、五和六个核苷酸,最小重复数分别为4、3、3、4和2。使用Primer Premier 5.0从差异基因表达数据中选择并设计这些SSR基因位点。SSR引物的参数设定为:GC含量40%~60%,引物长度20~25 bp,预期片段大小100~300 bp,退火温度55~65 ℃,上下游引物差异在2 ℃以内。
PCR扩增的反应体系为20 μL,包括:dd H2O 14.8 μL,dNTP 0.4 μL,缓冲液2 μL,20 μmol·L-1上下游引物各0.3 μL,DNA模板2 μL和Taq0.2 μL。PCR扩增的反应程序为:94 ℃预变性5 min;94 ℃变性30 s,54 ℃复性35 s,72 ℃延伸40 s,共35个循环;最后,72 ℃延伸3 min。通过1.5%琼脂糖电泳检测PCR扩增产物,点样量为3 μL。将3 μL可产生特定条带的PCR扩增产物与5 μL加载缓冲液混合。离心后,在94 ℃变性5 min,在冰箱中冷冻1 min,然后采用6%变性聚丙烯酰胺凝胶进行垂直电泳分析,银染后观察结果。能扩增出清晰条带的引物被视为有效引物,不能扩增出清晰条带或扩增条带不在范围内的引物被认为是无效引物。
2 结果与分析
2.1 转录组测序和组装
为了获得牡丹耐涝性的总体情况,使用高通量测序平台构建并测序了来自CK1、CK2、CK3(处理)和W1、W2、W3(对照)的6个cDNA文库。总共产生了444 382 372个Illumina原始读数,其长度范围从低于50 bp到151 bp。在去除接头序列和低质量读数后,获得了438 315 223个干净读数。每个样本中的表达基因数量在57 024到61 583之间。从组装转录物中共获得74 756个参考unigene,平均长度为743 bp,长度范围为201~12 252 bp。基于长度的unigene分布如图1所示。

图1 牡丹转录组unigene的长度分布和丰度Fig.1 Length distribution and abundance of unigenes from the P. suffruticosa transcriptome
2.2 功能注释
利用4个公共数据库(NR、Swissprot、KOG和KEGG)对unigene进行注释。注释结果显示,在NR数据库中有33 105个unigene(44.28%)匹配,在Swissprot数据库中有22 715个(30.38%)匹配,KOG数据库中有19 613个(26.24%)匹配,KEGG数据库中有12 763个(17.07%)匹配。共有33 278个unigene(44.51%)与4个数据库中的一个或多个数据库匹配,其中9 761个unigene在所有4个数据库中匹配(13.06%)。同时,这些结果表明,Illumina测序测试了牡丹中广泛表达的基因,其中6 106、1 986、1 964和1 921个分别与葡萄、核桃、可可和土瓶草的表达基因高度相似(图2)。
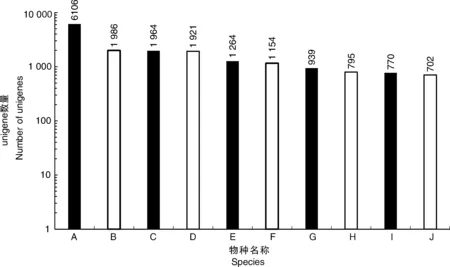
A,葡萄;B,核桃;C,可可;D,土瓶草; E,莲藕;F,酸枣;G,梅花;H,麻疯树;I,柑橘; J,木豆。A, Vitis vinifera; B, Juglans regia; C, Theobroma cacao; D, Cephalotus follicularis; E, Nelumbo nucifera; F, Ziziphus jujuba; G, Prunus mume; H, Jatropha curcas; I, Citrus sinensis; J, Cajanus cajan.图2 unigene BLASTX比对结果的物种分布图Fig.2 Species distribution of unigene BLASTX results
利用GO分析,将预测的牡丹unigene的功能分为3类:生物过程、细胞成分和分子功能。每个类别分别被进一步聚类为17、15和11个子类别,共包含43个子类别(图3)。对于生物过程类别,“细胞过程”(2 687)、“代谢过程”(3 297)和“单体过程”(2 163)是前3个富集组。细胞成分类别主要包含涉及“细胞”(1 588)、“细胞部分”(1 584)和“细胞器”(1 132)的基因。在分子功能类别中,“结合”(2 309)、“催化活性”(3 454)和“转运蛋白活性”(330)子类别的代表性很高。

a-q,生物过程(a,再生产;b,免疫系统过程;c,代谢过程;d,细胞过程;e,再生过程;f,生物黏附;g,信号传导;h,多细胞生物过程;i,发展过程;j,增长;k,单体过程;l,韵律过程;m,对刺激的反应;n,本地化;o,多生物过程;p,生物调控;q,细胞成分组织或生物发生); r-z,A-F,细胞成分 (r,细胞外区域;s,细胞;t,膜;u,病毒粒子;v,细胞连接;w,细胞外基质;x,膜封闭管腔;y,大分子复合物;z,细胞器;A,细胞外基质成分;B,细胞器部分;C,病毒粒子部分;D,膜部分;E,细胞部分;F,超分子纤维); G-Q,分子功能 (G,转录因子活性、蛋白质结合;H,核酸结合转录因子活性;I,催化活性;J,信号转换器活动;K,结构分子活性;L,转运蛋白活性;M,绑定;N,电子载体活性;O,抗氧化活性;P,分子转导活性;Q,分子功能调节剂)。a-q, Biological process (a, reproduction; b, immune system process; c, metabolic process; d, cellular process; e, reproductive process; f, biological adhesion; g, signaling; h, multicellular organismal process; I, developmental process; j, growth; k, single-organism process; l, rhythmic process; m, response to stimulus; n, localization; o, multi-organism process; p, biological regulation; q, cellular component organization or biogenesis) ; r-z, A-F, Cellular component (r, extracellular region; s, cell; t, membrane; u, virion; v, cell junction; w, extracellular matrix; x, membrane-enclosed lumen; y, macromolecular complex; z, organelle; A, extracellular matrix component; B, organelle part; C, virion part; D, membrane part; E, cell part; F, supramolecular fiber) ; G-Q, Molecular function (G, transcription factor activity, protein binding; H, nucleic acid binding transcription factor activity; I, catalytic activity; J, signal transducer activity; K, structural molecule activity; L, transporter activity; M, binding; N, electron carrier activity; O, antioxidant activity; P, molecular transducer activity; Q, molecular function regulator).图3 涝害胁迫下牡丹差异表达基因的GO分类图Fig.3 GO classification map of differentially expressed genes during waterlogging stress in P. suffruticosa
此外,共有32 059个unigene被分配到KOG分类中,并分为25个特定类别(图4)。最大的一组属于“仅一般功能预测”(7 512),其次是“翻译后修饰、蛋白质周转、伴侣蛋白”(3 604)和“信号转导机制”(2 901)。只有少数unigene被分为“细胞外结构”(68)和“细胞运动性”(13)。
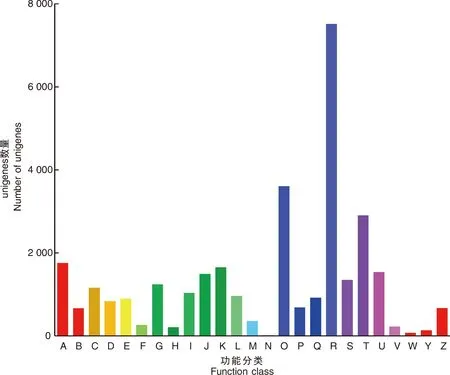
A,RNA加工和修饰;B,染色质结构和动力学;C,能源产生与转换;D,细胞周期控制、细胞分裂、染色体分割;E,氨基酸运输和代谢;F,核苷酸运输和代谢;G,碳水化合物运输和代谢;H,辅酶运输和代谢;I,脂质运输和代谢;J,翻译、核糖体结构与生物发生;K,转录;L,复制、重组和修复;M,细胞壁/膜/包膜生物发生;N,细胞运动性;O,翻译后修饰、蛋白质转运蛋白、伴侣蛋白;P,无机离子运输和代谢;Q,次生代谢产物的生物合成、运输和分解代谢;R,仅通用功能预测;S,功能未知;T,信号转导机制;U,细胞内运输、分泌和囊泡运输;V,防御机制;W,细胞外结构;Y,核结构;Z,细胞骨架。A, RNA processing and modification; B, Chromatin structure and dynamics; C, Energy production and conversion; D, Cell cycle control, cell division, chromosome partitioning; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; G, Carbohydrate transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; J, Translation, ribosomal structure and biogenesis; K, Transcription; L, Replication, recombination and repair; M, Cell wall/membrane/envelope biogenesis; N, Cell motility; O, Posttranslational modification, protein turnover, chaperones; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport and catabolism; R, General function prediction only; S, Function unknown; T, Signal transduction mechanisms; U, Intracellular trafficking, secretion, and vesicular transport; V, Defense mechanisms; W, Extracellular structures; Y, Nuclear structure; Z, Cytoskeletion.图4 涝害胁迫下牡丹差异表达基因的KOG分类和分析Fig.4 KOG classification and analysis of differentially expressed genes during waterlogging stress in P. suffruticosa
KEGG系统还对与涝害胁迫相关的unigene生物途径进行了注释。总共发现13 881个unigene参与了135个KEGG途径。涉及最多unigene的途径是“代谢途径”(2 733,39.22%),其次是“次级代谢产物的生物合成”(1 453,20.85%)和“抗生素的生物合成”(669,9.6%)。
2.3 涝害胁迫下的差异表达基因
为了可视化6个RNA-Seq样本之间的差异表达基因,进行了PCA绘图。该图显示,3个对照样品(CK1、CK2、CK3)和3个涝害处理的样品(W1、W2、W3)分别沿维度1和维度2清晰区分(图5)。为了鉴定在淹水胁迫下积累的牡丹的差异表达转录物,使用RPKM方法测量基因表达水平。使用FDR≤0.05和log2R≥1(R≥2)的值作为阈值来估计差异基因表达的显著性。在W组和CK组之间共检测到780个表达显著改变的基因,包括155个上调基因和625个下调基因(图6)。此外,在W和CK的文库中分别有71 187和70 697个转录本特异性表达。基于这些结果,我们发现牡丹的大多数基因在对淹水胁迫的反应中都有差异表达。在这些差异表达基因中,大量基因可能在淹水胁迫中发挥关键作用。
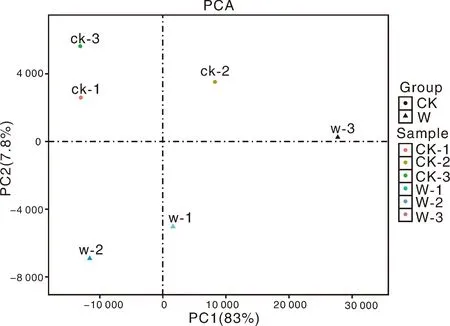
图5 六个RNA-Seq样本的主成分分析(PCA)图Fig.5 Principal component analysis (PCA) plot of 6 RNA-Seq samples
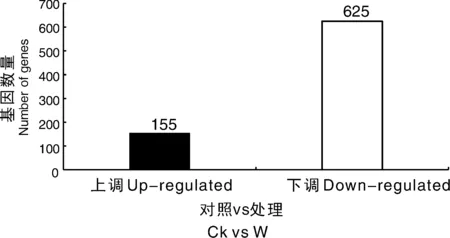
图6 涝渍胁迫下牡丹差异表达基因统计图Fig.6 Changes in gene expression profiles between the waterlogging stress and control libraries of P. suffruticosa
2.4 差异表达基因的功能分析
进行GO富集分析,对涝害处理组和对照组之间差异表达基因的推定功能进行分类。如上所述,差异表达的基因被分为3大类,包括生物进程、分子功能和细胞成分。在这3个类别下,前30个富集的功能组如表1所示,具体包括核小体组织、蛋白质-DNA复合物亚单位组织、染色体组织、蛋白质复合物亚单位组织等10个生物进程类功能组,蛋白质二聚化活性、蛋白质结合、核酸结合、杂环化合物结合等10个分子功能类功能组,以及无膜细胞器、细胞内无膜细胞器、核腔、细胞器腔等10个细胞成分类的功能组。

表1 GO类别下最富集的30个功能组Table 1 Top 30 most enriched functional groups under GO categories
为了进一步阐明差异表达的unigene在响应涝害胁迫中的功能,将差异表达的转录物与KEGG数据库类别进行匹配。结果显示,共有74条途径与涝害胁迫显著相关。前11个富集的功能途径如表2所示。在这11条KEGG途径中,3条最显著富集的途径分别是DNA复制、核糖体和嘧啶代谢。此外,还发现了与吞噬体、碱基切除修复、内质网蛋白质加工、嘌呤代谢等相关的其他重要途径,推测是牡丹响应涝害胁迫的主要途径。

表2 最富集的11个KEGG途径Table 2 Top 11 most enriched KEGG pathways
2.5 响应涝害胁迫的转录因子的鉴定
在涝害处理和对照条件下鉴定出多种差异表达的转录因子。在57个转录因子(TF)家族中,前10个TF家族如图7所示。在这10个TF家族中,大多数TF属于bHLH(92个unigene)、ERF(74个unigene)和MYB_related(65个unigene)。bHLH家族是在涝害胁迫条件下显著差异表达数量最多的一类转录因子,其他7个转录因子家族分别属于C2H2(62个unigenes)、FAR1(62个unigenes)、MYB(53个unigenes)、bZIP(51个unigenes)、C3H(50个unigenes)、NAC(44个unigenes)和WRKY(40个unigenes)家族。有25个、14个和12个差异表达的unigenes分别被分为HD-ZIP、ARF和AP2家族。
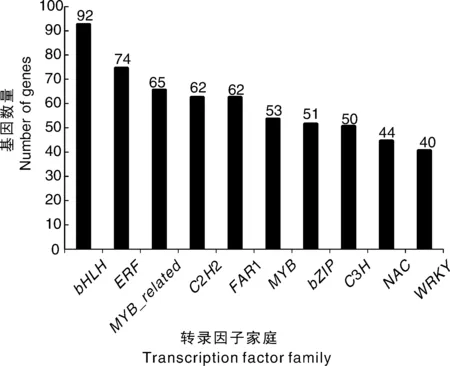
图7 前10个转录因子家族的基因数量Fig.7 Gene number of top 10 transcription factor families
2.6 qRT-PCR验证
为了验证RNA-Seq数据的准确性和可靠性,从差异表达基因中随机选择14个unigenes,使用特异性引物进行qRT-PCR分析。在14个被检测的unigenes中,11个基因的qRT-PCR表达模式与RNA-Seq数据(图8)显示出高度相似性。这一结果反映了基于RNA-Seq的差异表达分析是可靠和准确的。此外,使用倍数变化测量来计算RNA-Seq和qRT-PCR数据之间的相关性。随后,在两个数据组之间发现了良好的一致性(R2=0.933 8)(图8),证实了RNA-Seq方法获得的结果是有效和可靠的。
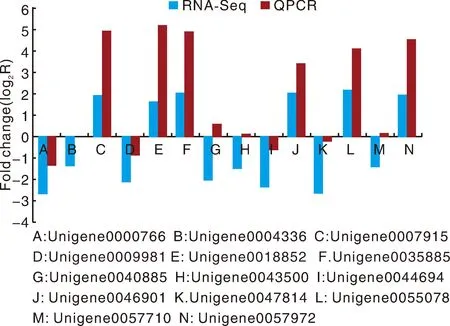
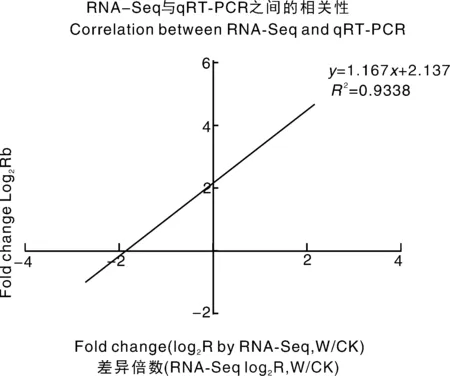
图8 qRT-PCR对基因相对表达水平的验证Fig.8 Verification of the relative expression levels of genes by qRT-PCR
2.7 SSR标记的分布和特征
从牡丹RNA-Seq数据中,在74 756个unigenes中发现了4 596个unigenes,包含5 204个SSR位点,频率为6.15%(包含SSR位点的unigene占总unigene的比例)。每个unigene至少有1个SSR位点,302个unigenes包含多个SSR位点。在5 204个SSR基因位点中,二核苷酸重复序列(3 025个,58.13%)占优势,其次是三核苷酸(1 411个,27.11%)、四核苷酸(377个,7.24%)、五核苷酸(128个,2.46%)和六核苷酸重复(263个,5.05%)(表3)。
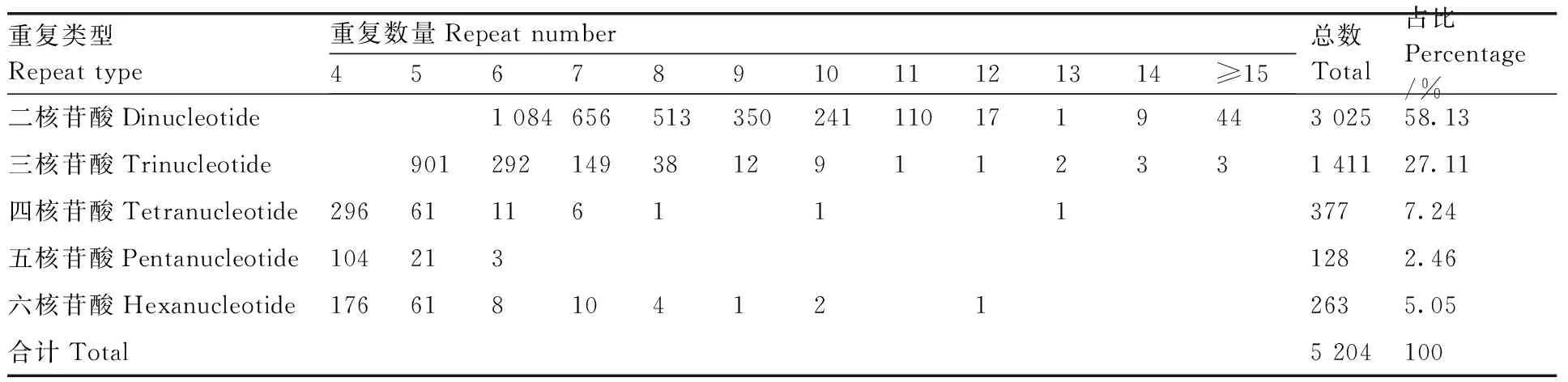
表3 牡丹耐涝相关unigene中SSR的类型、数量和分布频率Table 3 Type, number and distribution frequency of SSRs in Paeonia suffruticosa waterlogging related unigenes
对转录组中SSR的核苷酸类型进行分析,结果表明,在5 204个SSR位点中有212个重复基序。在二核苷酸重复基序中,最明显的显性基序是AG/CT(2 153,41.4%),其次是AT/AT(513,9.9%)和AC/CT(352,6.8%)。在三核苷酸重复基序中,最常见的基序是AAG/CTT(362,7%)、ACC/GGT(277,5.3%)和ATC/ATG(196,3.8%)。重复基序AAAT/ATT(171,3.3%)仅存在于四核苷酸重复基序中。然而,在其他类型中没有明显的显性基序(表4、图9)。
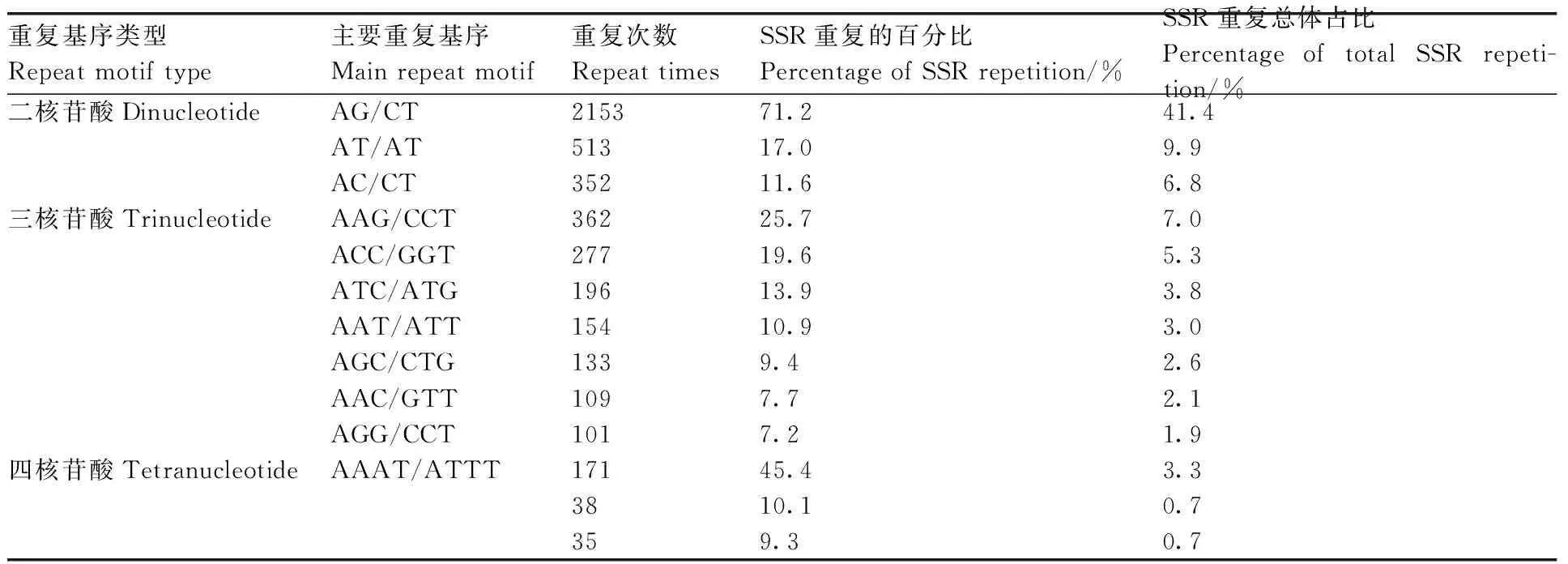
表4 牡丹涝害转录组SSR重复基序的类型和频率分布Table 4 Types and frequency distribution of SSR repeat motifs in the waterlogging transcriptome of Paeonia suffruticosa
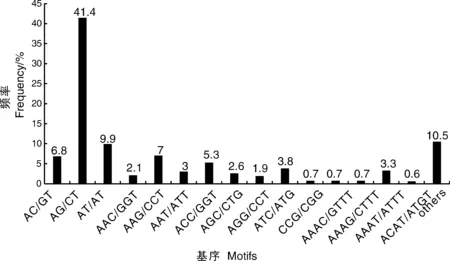
图9 二核苷酸、三核苷酸、四核苷酸和其他类型重复基序的频率分布Fig.9 Frequency distribution of di-, tri-, tetra-nucleotide and other type repeat motifs
2.8 涝害胁迫下SSR标记开发
本研究以与涝害胁迫调控相关的基因为候选基因,在叶片中发现了780个与涝害胁迫反应相关的差异表达基因。通过与SSR基因位点的比较,从差异表达的基因中发现55个含有SSR基因位点。设计了110对引物,筛选得到45对引物(表5)。以牡丹嫩叶DNA为模板,初步测试了引物的有效性,并对12对引物进行了二次筛选,在9个品种(红芙蓉、玉楼春、大富贵、一品红、岛锦、岛辉、凤丹和2个新品种)中扩增出清晰稳定的靶带(图10),平均多态性为83%。
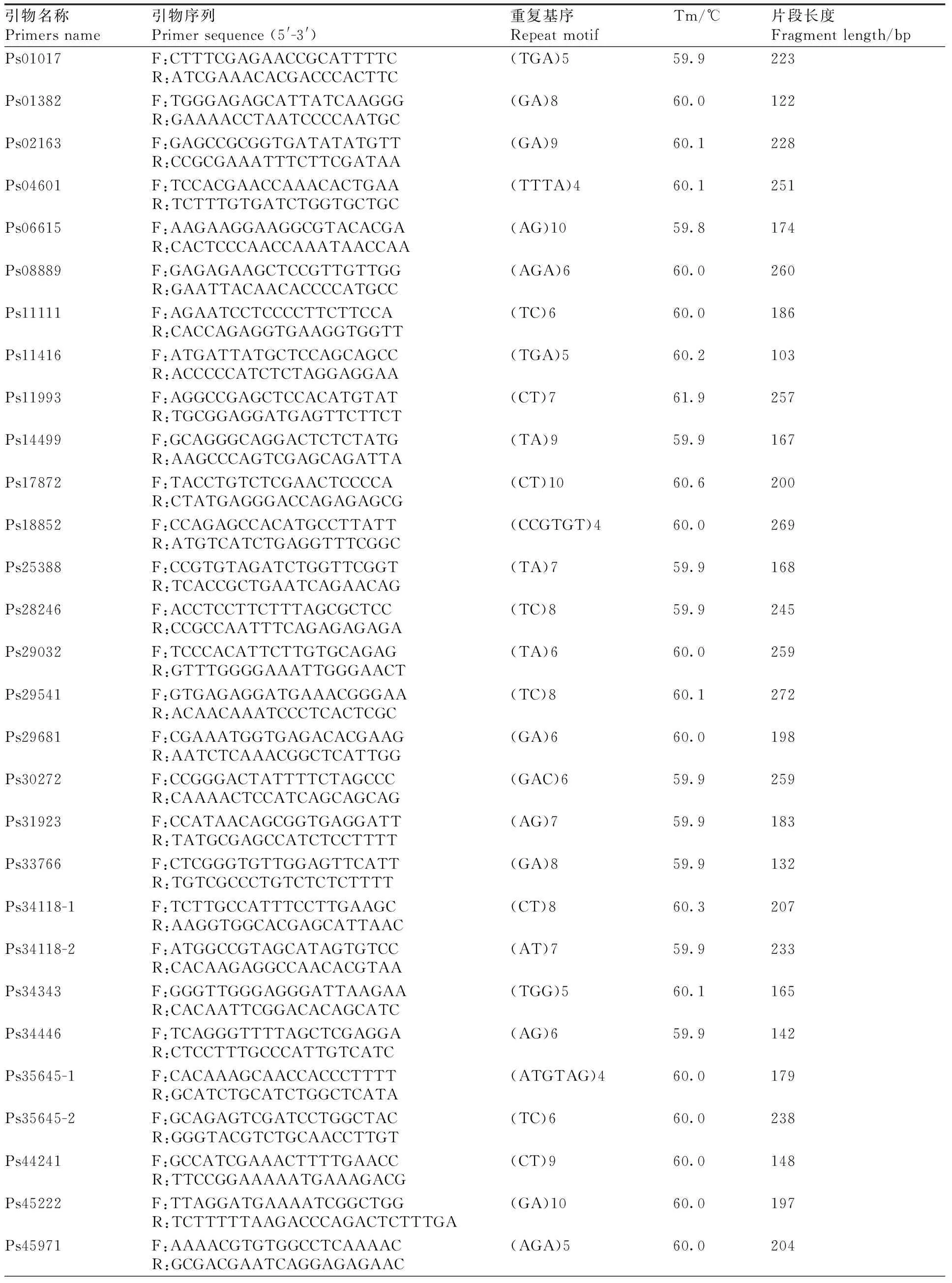
表5 牡丹涝害胁迫相关的SSR引物开发Table 5 SSR primers developed from genes related to waterlogging stress in Paeonia suffruticosa
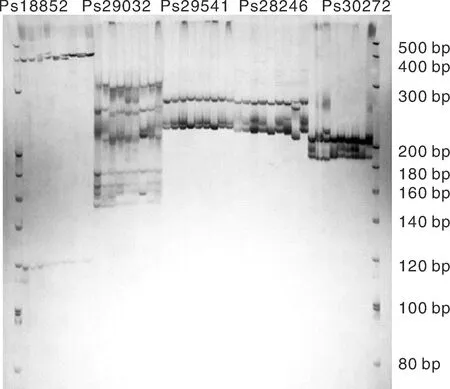
图10 SSR引物Ps18852, Ps28246, Ps29032, Ps29541和 Ps30272在9个牡丹品种中的扩增结果Fig.10 Polymorphism of Ps18852, Ps28246, Ps29032, Ps29541, and Ps30272 of SSR primers in 9 samples of P. suffruticosa
9个牡丹品种基因组DNA中12对引物的扩增带均具有多态性(表6),其中5对引物占100%的多态性,4对引物占80%以上的多态性。观察到的等位基因数(Na)在1.333 3~2.000 0,平均为1.830 7。有效等位基因数(Ne)在1.060 4~1.692 6,平均1.388 4。Nei基因多样性的变异范围为0.148 1~0.397 2,平均为0.256 1。香农信息指数在0.109 6~0.5581 8,平均值为0.372 0。这些结果表明,本研究开发的12对EST-SSR引物都是有效的通用引物,可用于牡丹品种的遗传多样性分析和品种鉴定。
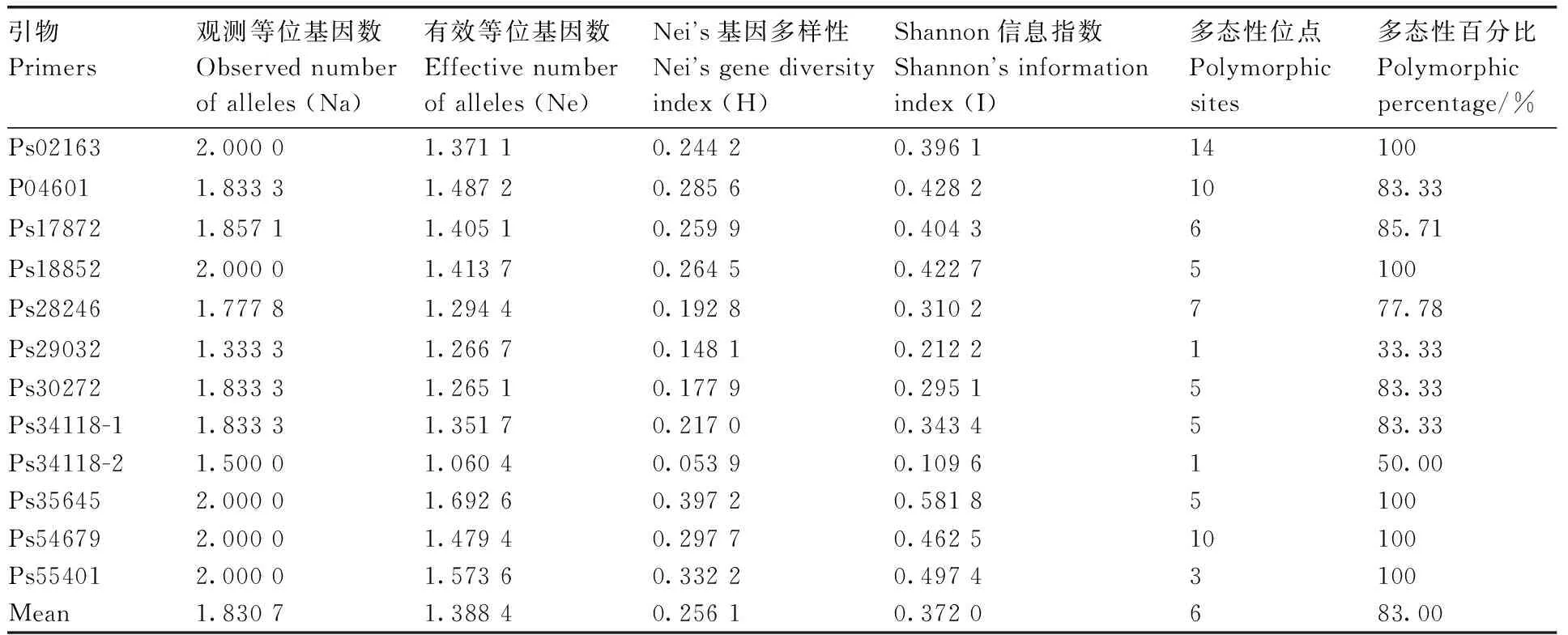
表6 十二个SSR引物在9个牡丹品种中的遗传多样性指数Table 6 Genetic diversity index for 12 SSR primers among 9 Paeonia suffruticosa cultivars
3 讨论
耐涝性是一个极其复杂的特征,涉及许多代谢途径和大量基因。在差异表达的基因中,一些具有功能注释的基因可能参与了耐涝性的调节,其中一些是未报道的基因。它们差异表达的原因和作用机制尚不清楚,需要进一步研究。对这些基因的进一步研究将有助于我们更好地了解牡丹耐涝性的调控机制。
本研究筛选出780个差异表达基因,其中37个差异表达最明显的基因与耐涝性相关,包含10个上调基因和27个下调基因。在37个差异表达基因中,有6个基因含有4个Hsp70和2个Hsp80,它们与热休克同源蛋白(Hsp)有关。Hsp70是一种热休克蛋白,长期以来一直被认为是最保守的蛋白家族之一。它可以对外部环境刺激作出反应,提高身体适应不利环境的能力。Hsp70蛋白广泛存在于细菌、植物和人类中[19]。在植物中,它们在应对非生物胁迫方面发挥着至关重要的作用,如热、冷、干旱和盐度等环境因素[20]。然而,Hsp对涝害胁迫的反应尚未报道。
通过分析,我们还发现了一些与转录因子相关的基因,如AP2/ERF、MYB和bZIP转录因子。这些转录因子均上调,并对其靶基因的表达产生进一步影响,从而决定牡丹的耐涝性。AP2/ERF基因家族成员被报道为与植物涝害胁迫相关的重要基因[21-23],ERFⅦ亚家族N末端的一个保守基序(NH2-MCGGAI/L)被证实与涝害胁迫适应有关[24]。MYB转录因子调节包括涝害胁迫在内的大量应激反应基因的表达谱[25-26]。例如,对棉花电子表达谱的分析表明,GhMYB基因在棉铃中特异性表达,并由根和叶中的耐涝胁迫诱导[27]。bZIP转录因子是近年来研究最多的转录因子家族之一。bZIP家族在包括拟南芥、烟草、水稻、小麦等在内的大量植物中被鉴定,其表达和功能被证明与耐涝性有关[25,28]。我们目前的研究结果进一步支持AP2/ERF、MYB和bZIP转录因子可能在牡丹的耐涝反应中发挥作用,并对许多耐涝基因的激活起重要作用,从而增强耐涝性。因此,提高一个关键转录因子的表达可能会提高牡丹的耐涝性。
还有一些其他与脱氢酶相关的蛋白质基因,如MDH、ADH、GDH等,以及一些编码渗透调节物质的相关基因如APX,可能在牡丹耐涝性的调节中发挥重要作用。
SSR标记作为一种能够区分显性和隐性等位基因的共显性标记,已被广泛用于研究种质之间的遗传关系和遗传多样性,以及连锁定位和基因定位[29-30]。新一代高通量测序技术(RNA-Seq)的快速发展为SSR标记的开发提供了一种快速有效的方法[31]。与普通的EST-SSR标记物相比,在基因或候选基因中开发的SSR可以更好地适应特定和多样化的市场需求。本研究在牡丹转录组数据库中检索了74 756个单基因拼接的SSR,获得了5 204个SSR位点,频率为6.56%,高于马尾松的4.32%[32]、菊花的2.84%[33],但低于三角梅的44.91%[34],24.46%的杜鹃[35]和15.2%的凤丹[14]。这表明转录组SSR中包含的信息不仅在物种之间不同,而且在品种之间也不同,或者SSR分析软件设定的标准不同。
多态性SSR分子标记的开发是物种重要性状分子标记辅助育种的基础和前提。在本研究中,获得了12个多态性引物,占有效引物的26.67%,低于牡丹(62%)[16],但高于牡丹(19.61%)[14]。它接近牡丹的27%[36]、25%[13]和24%[17]。大量研究表明,SSR基因座中重复序列的长度越长,SSR标记的多态性程度通常越高[37-38]。此外,多态性水平也可能受到遗传基础和实验材料样本量的影响[38]。例如,在24个芝麻样品中,多态性SSR的百分比为11.59%。样本量增加到25个后,多态性SSR的百分比上升到60.50%[39]。据预测,SSR引物多态性的比例也会随着样本遗传背景和样本量的增加而增加。
在本研究中,我们利用牡丹的转录组信息开发了多个与涝害胁迫基因相关的SSR分子标记,并验证了这些标记的有效性和普遍性。研究结果进一步丰富了牡丹的特异性分子标记数量,为后续牡丹分子标记辅助育种及相关分子生物学研究奠定了基础。接下来,我们将进一步验证这些SSR标记与耐涝性状之间的关系,为牡丹耐涝种质鉴定、遗传多样性分析和分子标记辅助育种提供科学依据。
