PPARγ低表达对AngⅡ诱导H9c2心肌细胞肥大的影响
2024-04-08赖玲缪林煜廖伟
赖玲 缪林煜 廖伟
(1赣州市人民医院,江西 赣州 341000;2赣南医科大学)
心血管疾病是全球发病率和死亡率的主要因素,也是许多慢性病的共同终点,而心肌肥厚是心力衰竭的一个主要病因。心肌肥厚有生理性和病理性两种类型。心肌肥大最初发展为对生理和病理刺激的适应性反应,但病理性心肌肥大通常进展为心力衰竭。每种形式的肥大都受到不同细胞信号通路的调节。目前,心肌细胞肥大(MCR)调控机制有脂肪代谢、胶原蛋白合成、增殖信号通路、转录因子。过氧化物酶体增殖物激活受体(PPAR)γ是过氧化物酶体增殖物配体激活后调节基因表达的转录因子之一。PPAR是心脏的生理主开关,指导心肌细胞的心脏能量代谢,从而影响心力衰竭、心肌肥厚。本研究旨在探讨PPARγ低表达对血管紧张素(Ang)Ⅱ诱导的H9c2 MCR的作用及可能机制。
1 材料与方法
1.1主要材料、试剂 大鼠心肌细胞株H9c2购自中国科学院细胞库;AD-PPARγRNAi腺病毒购于上海吉凯基因;PPARγ、肌球蛋白重链(MYH)7B、心钠肽(ANP)抗体购于Abcam 公司;抗小鼠IgG-辣根过氧化物酶(HRP)、抗兔IgG-HRP二抗购自Abclonal公司;β-actin 小鼠单克隆抗体购自中山金桥公司;AngⅡ购自Abcam 公司;胎牛血清购自天津灏洋生物制品科技有限责任公司;DMEM、胰酶、磷酸盐缓冲液(PBS)、二甲基亚砜(DMSO)、RIPA 裂解液购自索莱宝公司;二喹啉甲酸(BCA)蛋白定量试剂盒购自碧云天;超敏化学发光试剂盒购自Thermo公司。
1.2细胞培养 用含10%胎牛血清的DMEM 培养液培养细胞,隔天换液1次,待细胞长至约占培养皿90%时,用PBS 冲洗2遍,加数滴25%胰酶,消化1~2 min,加含血清培养液终止消化,用枪头轻轻吹打使细胞脱落,离心、重悬后按1∶3比例接种到新的培养皿中。
1.3构建MCR模型 把需要用的实验材料及试剂放于细胞房无菌超净台桌面上打开紫外灯,消毒30 min。在超净工作台将旧培养基倒入废弃桶,PBS洗涤2次,然后用移液管吸干剩余的培养溶液。最后加入新配浓度为10-7mol/L的AngⅡ培养基,做好标记,继续放入孵箱培养至48 h。
1.4细胞分组 采用AD-PPARγRNAi重组腺病毒载体,感染到H9c2心肌细胞中。最终分为对照组、Vehicle组、AD-PPARγRNAi组;MCR组、MCR+Vehicle组、MCR+AD-PPARγRNAi组6组。
1.5AD-PPARγRNAi感染H9c2细胞 在病毒房细胞超净台内,准备好细胞换液的实验用品,打开紫外线照射30 min,同时打开病毒房紫外线照射30 min。将细胞板中的液体倒入台面上的废弃桶,同时加入5 ml PBS,稍微摇晃几下,将液体倒入台面上的废弃桶,PBS洗涤2次。将计划好的病毒液体与培养液混匀后分别加入到Vehicle组、AD-PPARγRNAi组、MCR+Vehicle组、MCR+AD-PPARγRNAi组的细胞板内,做好标记,放置孵箱最底层,培养2 h后,再次增加5 ml培养液。培养12 h后,将含病毒液培养的 H9c2细胞从孵箱中取出,将细胞在病毒室的洁净室中更换,并继续在培养箱中放置48 h。
1.6H9c2细胞苏木素-伊红(HE)染色 (1)细胞状态达90%左右时可进行HE染色。6组均需要PBS洗涤2次;取出储存在4 ℃冰箱中的4%多聚甲醛,均匀加入细胞中固定10 min。PBS洗涤3~5次,将移液枪吸取残留余PBS并干燥,均匀加入苏木素染液浸染8 min,分别加入二蒸水浸洗数遍,控干水分。加入分化液分色数秒,再蓝化H9c2细胞的核通过用准备好的凉水(100 ℃水放凉后)。用移液管吸取伊红染液在细胞板内,5 min,在水龙头下直接冲洗。最后脱色透明后在镜下观察拍照,每组重复3次。
1.7Western印迹法检测蛋白表达
1.7.1细胞蛋白提取 (1)配制细胞裂解液:计算好需要的量并预先冷却RIPA 裂解液,PMSF按100∶1比例准备,将其放在涡流振荡器上并充分混合,放在冰上或保存在4 ℃的冰箱中。(2)将细胞板中的液体倒入废弃桶,同时加入5 ml PBS,稍微摇晃几下,将液体倒入废弃桶,重复再洗1次后,分别用大、中、小移液枪反复将剩余液体吸干净,尽量去除剩余液体。(3)用移液枪吸取300~400 μl预冷后的细胞裂解液均匀滴加在培养皿中,放入4 ℃冰箱或冰上,5 min后轻轻摇晃使裂解液尽量覆盖细胞上,继续裂解5 min。(4)首先把细胞板放在冰上,接着用消毒后的细胞刮在细胞板上将H9c2细胞全部刮下至细胞板面光滑为止,最后将液体转入离心管内。(5)将含有细胞液的离心管用透明薄膜密封,置于4 ℃离心机中,以12 000 r/min离心15 min,最后保留上清液用于标记。(6)再将含蛋白质且标记好的EP管封口处理后存放在-20 ℃冰箱,不要反复冻融。
1.7.2蛋白浓度测定 (1)配制工作液:计算好需要的工作液,按混合试剂A与试剂B为50∶1比例配制,放于旋涡振荡器上充分混匀后,将其放置冰上或4 ℃冰箱保存待用。(2)从-20 ℃冰箱内取出BSA标准液室温下溶解,配制标准品蛋白溶液:吸取20 μl BSA至EP管,再加入30 μl ddH2O,混匀,从中取出25 μl连续倍比稀释,标准浓度为:0、25、50、100、200、400、800、1 600 μg/ml,在96孔板中向每个孔中加入25 μl。(3)待测样品处理:取每组样品用三蒸水分别稀释成0、1、5、10、20倍,依次加入25 μl至96孔板中。(4)将配好的BCA工作液分别加入200 μl至标准蛋白液孔中及待测样品空中,轻柔混匀,放于37 ℃恒温箱,30 min。(5)提前打开酶标仪,预热。将96孔板放置在酶标仪的指定位置,设置A562 nm的波长,运行程序,记录光密度(OD)值,并根据每个孔测量的数据获得标准曲线,从而计算样品上样量。
1.7.3电泳分离 (1)洗干净配胶板同时用ddH2O冲洗,并固定在配胶固定架上,用橡胶板注入ddH2O泄漏检测5 min,并去除板表面之间的 ddH2O备用。(2)接着按照表1分别配制12%、10%、8%的分离胶。(3)放在振荡器上混匀后,用大移液枪迅速将液体加入玻璃板间隙内,操作过程中注意不要产生气泡,接着加入1.5 ml异丙醇排除气泡在室温下放置50 min,倒掉异丙醇同时用ddH2O洗干净,并用滤纸吸干净。(4)配制5%聚成胶:其中ddH2O 3.4 ml,30% Acr-Bic 0.83 ml,Tris.HCl(pH6.8),0.63 ml 10%SDS 0.05 ml,10%AP 0.05 ml,TEMED 0.005 ml。(5)放在振荡器上混匀后,用大移液枪迅速将液体加入玻璃板间隙内操作过程中注意不要产生气泡,同时将晾干的梳子快速的插入,室温条件下放置45 min左右。(6)上样:根据BCA法蛋白定量结果及实验分组调整进样量以使每个泳道蛋白上样量一致。用进样针吸取相应体积的蛋白缓慢注入泳道内,并在两侧分别注入3~5 μl蛋白Marker。(7)电泳槽被固定好后,并注入现配好的1倍电泳缓冲液,插好电泳仪电源,设置90 V恒压30 min条件,当溴酚蓝到达分离层时,将其调到120 V恒压,且电泳持续60 min至溴酚蓝到底部。

表1 分离胶配制
1.7.4蛋白的膜转移、显色、检测 (1)湿转:从电泳槽中取出胶板,按蛋白分子量大小,参照蛋白分子 marker,用刀片切下相应的胶块,浸泡在预冷的1倍湿转缓冲液中,将聚偏氟乙烯(PVDF)膜在甲醇中电离1 min,制备膜夹。按照“黑胶白膜”的原则放入切好的胶块及 PVDF膜,排尽膜与胶之间的气体,堵住湿转移夹,盖住电转移装置,调好湿转移条件,四周放入冰水及冰袋。(2)封闭:从湿转移装置中取出PVDF膜,轻轻冲洗TTBS并置于现成的5%脱脂乳中,在室温下封闭2 h。(3)一抗孵育:从封闭溶液中取出PVDF膜,轻轻冲洗TTBS以除去残留的封闭溶液,然后置于相应的一抗中,并在4 ℃温育过夜。(4)二抗孵育:第2天,取出PVDF膜,漂洗TIBS 10 min×4(根据暴露背景适当调整特定的漂洗时间),根据初级来源选择二级抗体。(5)曝光:二抗中取出PVDF膜后,TTBS漂洗10 min×5(具体漂洗时间根据曝光背景做出适当调整),吸干PVDF膜上残留TBS,将ECL试剂盒中A与B溶液以1∶1比列混匀后,把PVDF膜放在混合液中,最后曝光成像同时保存图片。
1.8统计学方法 采用SPSS22.0软件进行方差分析、LSD-t检验。
2 结 果
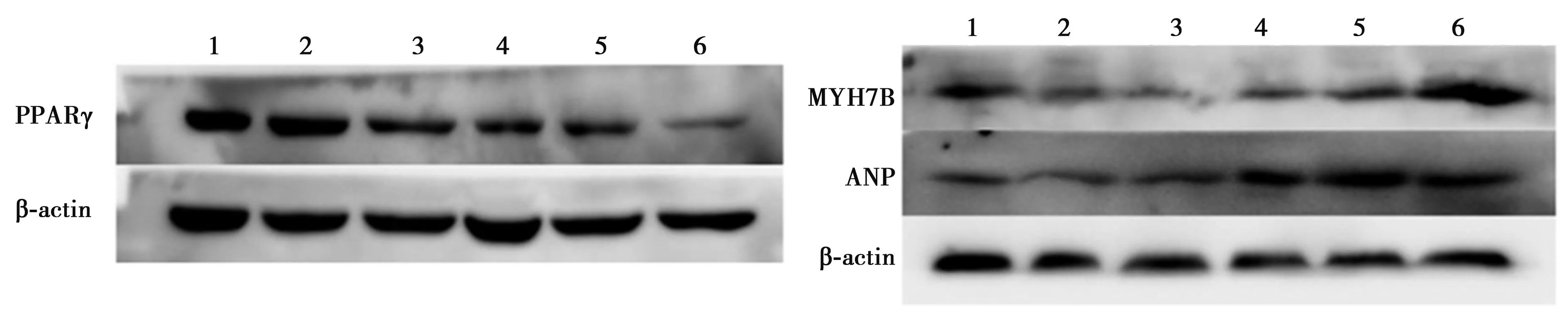
2.1H9c2细胞模型的构建 HE染色结果显示,MCR组与对照组比较,H9c2细胞核形态大小变大,见图1。Western 印迹结果显示:与对照组比较,MCR组MYH7B、ANP的表达显著增加(P<0.05),表明MCR模型构建成功。见图2、表2。
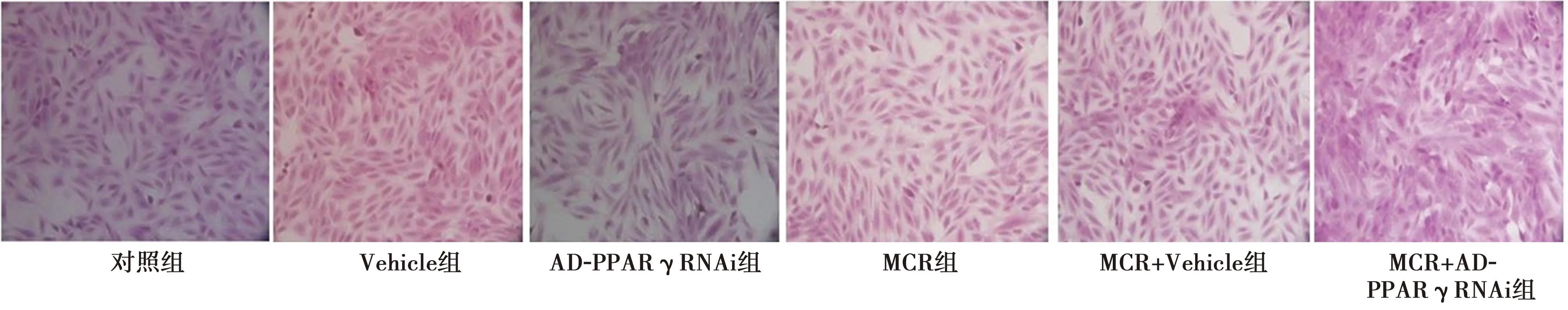
图1 HE染色H9c2细胞(×200)
1~6:对照组、Vehicle组、AD-PPARγRNA组、MCR组、MCR+Vehicle组、MCR+AD-PPARγRNAi组

表2 AD-PPARγRNAi感染H9c2细胞PPARγ、MYH7B、ANP的相对表达量
2.2各组PPARγ低表达比较 AD-PPARγRNAi组较对照组和Vehicle组PPARγ表达明显降低(P<0.05);MCR+AD-PPARγRNAi组较MCR和MCR+Vehicle组明显降低(P<0.05),见表2。
2.3空白对照与病毒对照比较 HE染色结果显示,Vehicle组较对照组H9c2细胞核形态大小无明显变化,MCR+Vehicle组与MCR组比较H9c2细胞核形态大小无明显变化;Vehicle组与对照组比较、MCR+Vehicle组与MCR组比较PPARγ、MYH7B、ANP表达均无明显差异(P>0.05),见图1、图2、表2。
2.4AD-PPARγRNAi感染H9c2细胞MYH7B、ANP的相对表达情况比较 Western印迹条带分析显示,与对照组比较,AD-PPARγRNAi组MYH7B、ANP表达明显增加(P<0.05);与MCR组比较,MCR+AD-PPARγRNAi组MYH7B、ANP表达明显增加(P<0.05),见表2。
3 讨 论
心肌肥厚是导致心力衰竭的一个重要病因,也会导致心脏疾病患者死亡。病理性心肌肥大除了细胞生长和蛋白质合成外,心肌肥大会变成适应不良的失代偿:细胞死亡、纤维化、Ca2+处理蛋白失调、线粒体功能障碍、代谢重塑、胎儿基因表达重新激活、蛋白质和线粒体调控受损、肌节结构改变和血管生成不足。诱导这些反应的信号机制促进了适应不良的心脏重塑和功能障碍,并最终导致心力衰竭。因此,伴随上述反应的心肌肥大通常被认为是病理性的。抑制心肌肥大的信号通路在治疗上可能存在重要作用。高血压和心肌梗死等病理状况主要通过神经内分泌激素和机械力促进病理性肥大,并伴有与生理性肥大不同的信号通路。
心脏代谢在生理和病理性心肌肥大中受到不同调节〔1〕。全身及心脏代谢的变化在心力衰竭发展之前〔2,3〕。心肌肥大反应过程中能量代谢的适应受损加剧病理性心肌肥大,增加心肌细胞死亡。ATP产生的过程直接改变收缩功能并导致心力衰竭〔4,5〕。在心肌肥大过程中,雌激素受体相关受体(ERR)α与过氧化物酶体增殖物激活受体-γ共激活因子(PGC)1α相互作用,通过增加编码参与三羧酸(TCA)循环的蛋白质基因表达来激活线粒体能量代谢和氧化磷酸化。尽管编码ERRα的基因缺失不会影响TAC诱导心肌肥大的最初发展,但ERRα缺乏会促进向心力衰竭的转变〔6〕。编码清道夫受体基因的心脏特异性CD36缺失,将减少心脏中脂肪酸的摄取和利用,加剧压力超负荷引起的心功能不全和心力衰竭,但不影响心肌肥大程度〔7〕。以上表明适当的代谢作用将适应病理性心肌肥大维持收缩功能。编码胰岛素受体基因的心脏特异性缺失通过损害脂肪酸和丙酮酸代谢及 TCA 来促进线粒体功能障碍、氧化应激和收缩功能障碍〔8〕。因此,诱导生理性心肌肥大的信号机制可能协调线粒体的代谢来维持心脏功能。改变的代谢物也会促进病理性心肌肥大或心力衰竭。基因工程小鼠模型中脂肪酸或碳水化合物代谢失调直接诱导心肌肥大或通过调节病理性心肌肥大的信号机制导致功能下降〔9~14〕。
PPAR是一类配体激活的核转录因子在脂质和脂蛋白代谢、葡萄糖平衡中的作用代谢、细胞增殖、分化和凋亡中发挥重要作用。根据专业结构,PPARs可分为3个亚组:PPARα〔也称为核受体亚家族 1 组 C 成员(NR1C)1〕、PPARδ(也称为 PPARβ 或 NR1C2)和 PPARγ(也称为 NR1C3)。最初发现,PPARγ主要是在脂肪细胞中调节脂肪细胞的分化而脂质代谢和提高胰岛素敏感性,故与高血压、肥胖、动脉粥样硬化、Ⅱ型糖尿病、胰岛素抵抗密切相关。最近发现,PPARγ也在心脏、血管平滑肌细胞、内皮细胞等部位中表达。此外,PPARγ激活剂吡格列酮和罗格列酮可抑制AngⅡ诱导的新生大鼠MCR。表明PPARγ参与了MCR的病理生理过程。
本研究通过高糖培养基结合AngⅡ培养H9c2 心肌细胞48 h MYH7B、ANP表达增加,再给予AD-PPARγRNAi重组腺病毒感染后MYH7B、ANP表达上调。
综上,保护心肌细胞中PPARγ至关重要,后续研究继续探讨PPARγ高表达是否对改善或逆转MCR有重要作用,本研究为了解心肌肥厚机制、心肌细胞的防治策略及以PPARγ为靶点新药开发提供依据。
