特发性肺纤维化和炎症性肠病共同核心基因特征及其生物学机制的鉴定
2024-04-08娄娇娇王浩冉黄凤祥王焕勤康燕王起龙马开乔瑞萍苗丽君
娄娇娇,王浩冉,黄凤祥,王焕勤,康燕,王起龙,马开,乔瑞萍,苗丽君
(郑州大学第一附属医院 呼吸与危重症医学科,河南 郑州 450052)
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是一种慢性、致死性间质性肺疾病,中位生存期为3~5 a[1]。IPF的诊断需要在合适的临床条件下通过高分辨率CT(high-resolution computed tomography,HRCT)或组织学检查发现典型的间质性肺炎,且缺乏明确的病因[2]。炎症性肠病(inflammatory bowel disease,IBD)是一种描述多种胃肠道炎症性疾病的广义术语,主要类型是克罗恩病(Crohn’s disease,CD)和溃疡性结肠炎(ulcerative colitis,UC)[3]。IBD在肺部的肠外表现包括上气道疾病伴狭窄、大气道疾病伴炎症、支气管扩张、炎症性小气道疾病、肺实质疾病、血管疾病、间质性肺疾病、肉芽肿性肺疾病和嗜酸性肺炎[4-6]。
有研究表明IBD与IPF相关,IBD患者发生IPF的风险更高[7-8]。尽管近年来IBD与IPF的相关性受到相当多的关注,但相关研究仍较少,需要进一步探索。本研究的目的是通过生物信息学分析确定与IPF和IBD相关的核心基因。对核心基因进行了富集分析,并进一步分析了与这些基因相关的转录因子(transcription factors,TFs)及微小核糖核酸(microRNAs, miRNAs),旨在探索IPF和IBD相似的遗传特征和潜在生物学机制,为进一步研究IPF和IBD共同发病机制提供新的研究方向。
1 资料与方法
1.1 资料下载
通过公共开放的基因表达综合数据库(gene expression omnibus,GEO)(http://h-p.www.ncbi.nlm.nih.gov.zzulib.vpn358.com/gds/)筛选了与IPF和IBD相关的转录组测序数据集,其中IPF的基因表达谱芯片数据集为GSE53845和GSE110147,IBD基因表达谱芯片数据集为GSE59071和GSE75214。使用GSE110147和GSE75214筛选差异表达基因(differentially expressed genes,DEGs),使用GSE53845和GSE59071进行枢纽基因表达验证。
1.2 差异表达基因筛选
首先,根据对应平台中对数据集的注释文档,使用R软件(4.2.1)“merge”包将获得的4个数据集中的探针与基因符号进行匹配。然后,利用R中的“limma”包对4个数据集进行背景校准、归一化和log2转换。当多个探针对应同一基因时,计算平均值以确定其表达量。对于GSE110147和GSE75214,使用R中的“limma包”通过设置条件截断标准|log2FC|>1和P<0.05来筛选DEGs。使用R软件制作热图和火山图来可视化这些DEGs。将IPF和IBD的DEGs取交集,得到共同的DEGs,通过构建维恩图显示重叠的DEGs。
1.3 差异基因的功能富集分析
研究通过使用“clusterProfiler”包和“GOplot”包对DEGs进行基因本体论(gene ontology,GO)富集分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)富集分析明确共同的DEGs的潜在生物学功能。
1.4 蛋白质-蛋白质相互作用网络的构建及核心基因的鉴定
STRING是一个利用实验数据和计算预测方法预测蛋白质相互作用的数据库。此研究使用STRING(http://string-db.org.zzulib.vpn358.com/)构建了常见DEGs的蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络。将置信度评分设置为>0.4,隐藏网络中断开的节点。利用Cytoscape(3.9.1)的CytoHubba和MCODE功能PPI网络复合体中发现关键基因。MCODE默认设置;在CytoHubba中从12种算法中随机选取5种算法计算前15个关键基因并且通过在线维恩图工具(http://h-p.bioinformatics.psb.ugent.be.zzulib.vpn358.com/webtools/Venn/)将5种算法的运行的结果取交集确定核心基因。
1.5 核心基因表达的验证
使用IPF数据集GSE53845和IBD数据集GSE59071进一步验证这些核心基因在两种疾病中的表达水平,差异表达水平的比较采用t检验,以P<0.05为差异有统计学意义。通过R软件“ggpubr”包获得的小提琴图谱显示了不同组间核心基因的表达水平。对核心基因进行GO富集分析和KEGG分析。
1.6 TFs-核心基因相互作用构建及TFs-miRNAs-核心基因网络预测
TFs是基因表达调控中的关键分子,可以与特定的DNA序列结合。JASPAR(http://h-p.jaspar.genereg.net.zzulib.vpn358.com)是一个开放访问的TFs数据库,TFs结合图谱存储6个分类群的多个物种的TFs结合位点频率矩阵。使用Networkanalyst工具(version 3.0, http://www.networkanalyst.ca.zzulib.vpn358.com/)评估JASPAR数据库中TFs基因与IPF和IBD共同的核心基因的交互作用。miRNAs可通过与靶mRNA结合来调控靶基因的表达。了解TFs与miRNAs之间的调控转录网络将有助于研究不同生理和疾病条件下基因表达的机制。使用NetworkAnalyst 3.0软件分析TFs-miRNAs共调控网络,并用Cytoscape软件对结果进行可视化分析。
2 结果
2.1 IPF和IBD中共同的DEGs
将GSE110147数据集中的22例IPF样本与11例正常对照进行比较,得出3 370个DEGs。将GSE752141数据集中的172例IBD样本与22例正常对照进行比较,筛选出了460个DEGs。通过火山图和热图对2个数据集的DEGs进行可视化(图1)。此外,利用维恩图对GSE110147和GSE752141之间的共同DEGs进行分析。结果显示,在这2个数据集中,有90个基因的表达发生了显著变化,其中上调基因67个,下调基因23个。

A为GSE110147 DEGs的火山图;B为GSE752141 DEGs的火山图;C为GSE110147 DEGs的热图;D为GSE752141 DEGs的热图。
2.2 IPF和IBD中共同的DEGs的GO和KEGG富集分析
使用R软件中的“cluster profiler”包对这些共同的DEGs进行GO分析和KEGG通路富集,进一步探索潜在的生物信息。分析结果表明,在生物过程(biological process,BP)方面,这些基因主要与生长、细胞外基质组织、细胞外结构组织的负调控相关。在细胞成分(cellular component,CC)方面,基因主要与内质网管腔、含胶原的细胞外基质相关。最后,在分子功能(molecular function,MF)方面,这些基因主要与细胞外基质结构成分、内肽酶活性相关(图2A、2B)。此外,KEGG分析显示,DEGs主要富集于黏着斑、人乳头瘤病毒感染、PI3K-Akt信号通路、ECM-受体相互作用、矿物质吸收(图2C、2D)。

A为常见DGEs的GO富集分析条形图;B为常见DGEs的KEGG富集分析柱状图;C为常见DGEs的GO富集分析气泡图;D为常见DGEs的KEGG富集分析气泡图。
2.3 PPI网络构建与核心基因鉴定
为了进一步探索共同DEGs编码的蛋白质之间的潜在相互作用,并识别核心基因,此研究利用STRING数据库对DEGs进行了PPI网络分析(图3A)。使用Cytoscape 3.9.1软件进行网络分析和可视化。CytoHubba插件通过5种算法得到7个基因(SPP1、COL1A1、POSTN、MMP1、MMP7、COL3A1、COL6A3)(图3B)。然后,应用MCODE插件进行模块分析(过滤标准:degree cut-off=2;node score cut-off=0.2;k-core=2;max depth=100),在网络中得到4个模块,如图3C~G所示。将CytoHubba获得的基因与MCODE获得的基因取交集,得到6个枢纽基因:SPP1、COL1A1、POSTN、MMP7、COL3A1、COL6A3。
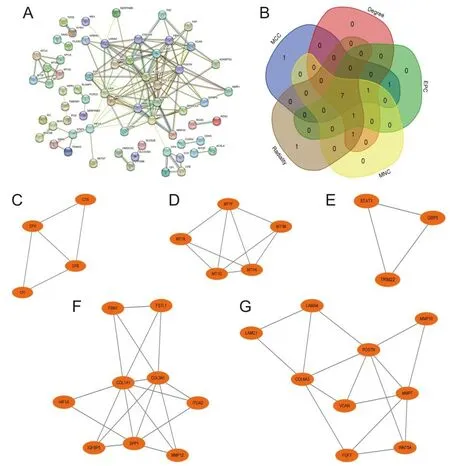
A为STRING构建的常用DEGs的PPI网络;B为通过5种算法鉴定的7个候选基因;C~F为显著性基因模块及模块基因。
2.4 核心基因的验证
使用另外2个数据集,IPF的GSE53845和IBD的GSE59071分析核心基因的表达水平对核心基因表达水平的置信度进行验证。结果显示,在GSE53845中,与健康组相比,IPF组的所有核心基因均显著上调(图4A)。同样,在GSE59071中,与健康组相比,IBD组的所有核心基因均显著上调(图4B)。

A为GSE110147数据集中IPF组与对照组的核心基因表达;B为GSE75214数据集中IBD组与对照组的核心基因表达;*P<0.05; **P<0.01;***P<0.001;ns为P>0.05。
2.5 核心基因的GO和KEGG富集分析
GO富集分析显示,枢纽基因主要集中在细胞外基质组织、细胞外结构组织、内质网管腔、含胶原的细胞外基质、细胞外基质结构成分。KEGG富集分析显示,枢纽基因主要集中于ECM-受体相互作用、黏着斑、人乳头瘤病毒感染、PI3K-Akt信号通路(图5)。
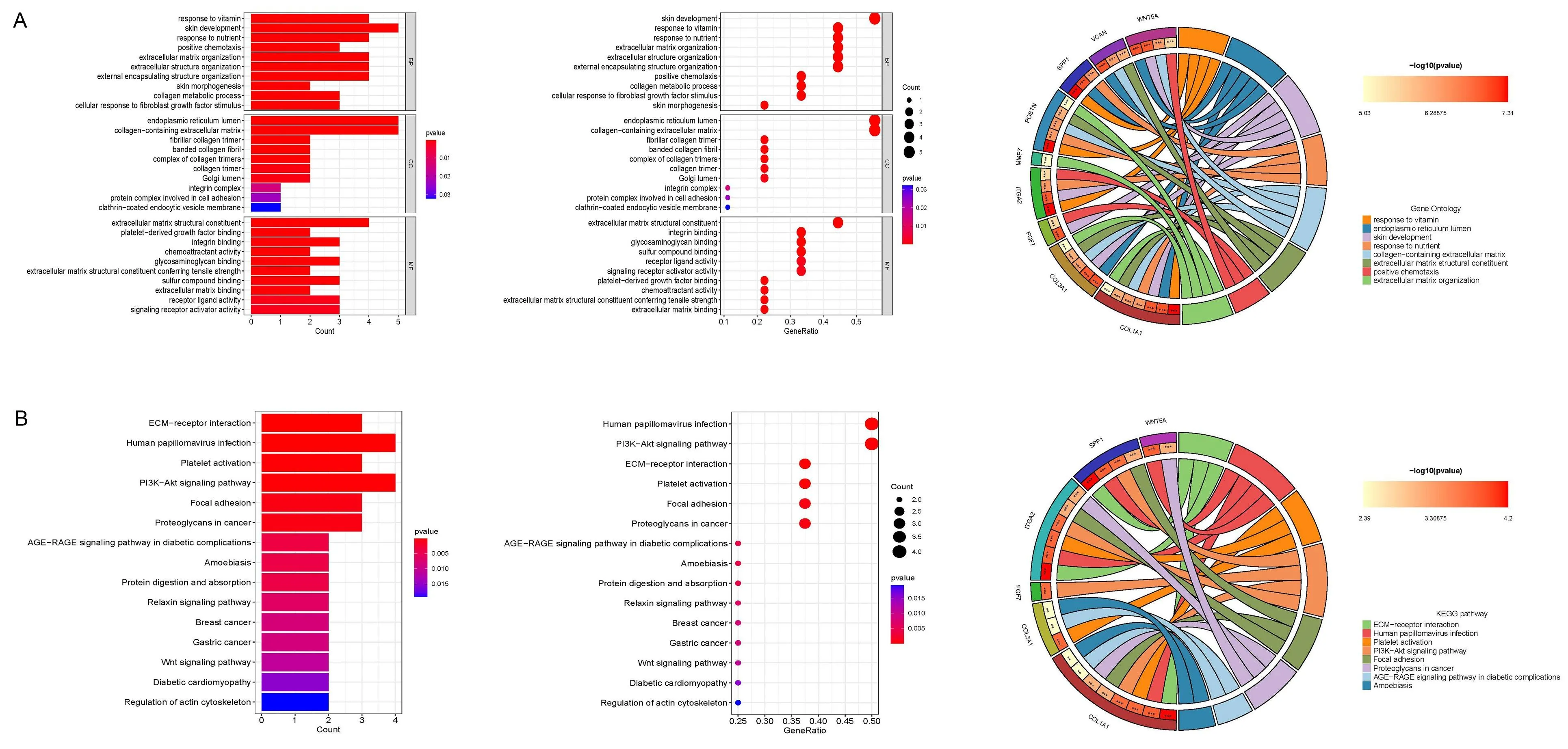
A为核心差异基因GO富集分析的条形图、气泡图和圆图;B为核心差异基因KEGG富集分析的条形图、气泡图和圆图。
2.6 核心基因-TFs和核心基因-miRNAs相互作用网络
对于核心基因,通过NetworkAnalyst平台构建了包含25个交互作用、32个TFs和6个核心基因的TFs调控网络(图6A)。TFs-miRNAs调控网络揭示了miRNAs和TFs之间的关系以及与核心基因的相互作用。通过NetworkAnalyst构建TFs-miRNAs调控网络,通过Cytoscape的CytoHubba插件的MCC算法得到85个miRNAs和34个TFs(图6B)。

A为TFs-核心基因网络图;B为TFs-miRNAs调控网络网络图。
3 讨论
IPF是一种以成纤维细胞向肌成纤维细胞转化和细胞外基质过度沉积为特征的慢性进行性纤维化肺疾病[9]。IBD被越来越多的认为是一种复杂的疾病,可由多种原因引起或加重,在一般人群中的发病率呈上升趋势[10]。IPF与慢性肝炎、原发性胆汁性肝硬化、EB病毒感染、IBD、Whipple病相关[11]。然而,目前很少有研究从基因水平探讨IPF和IBD的共同发病机制,因此本研究旨在探讨IPF与IBD之间的关系。研究使用来自IPF(GSE110147)和IBD(GSE752141)的样本数据集获得了90个共同的DEGs。基于Cytoscape的MCODE插件和CytoHubba插件,在PPI网络中筛选出6个重叠的DEGs作为核心基因,包括SPP1、COL1A1、POSTN、MMP7、COL3A1、COL6A3。这6个基因在IPF和IBD患者中均表达上调,提示这些基因可能在IPF和IBD的发病机制中发挥重要作用。GO富集分析结果显示,枢纽基因主要富集于细胞外基质组织、细胞外结构组织、内质网管腔、含胶原的细胞外基质、细胞外基质结构成分。KEGG富集分析显示,核心基因主要集中在ECM-受体相互作用、黏着斑、人乳头瘤病毒感染、PI3K-Akt信号通路。其中,ECM-受体相互作用和黏着斑已被证实与IPF的调控密切相关,PI3K-Akt通路通过其促炎作用和活化T细胞参与IBD的发病[12-14]。
分泌型磷蛋白1(secreted phosphoprotein 1,SPP1)又称骨桥蛋白(osteopontin,OPN)样蛋白,是一种多功能蛋白,表达于活化的巨噬细胞、T细胞、破骨细胞、肝细胞、平滑肌、内皮细胞和上皮细胞,可介导细胞黏附、趋化、信号转导和组织修复等多种生物学功能[15-17]。SPP1/OPN是IPF的标志物,在人类IPF中表达上调,对人类IPF起促进作用[18-19]。既往研究表明,SPP1/OPN在IBD患者肠上皮细胞和炎症肠组织中浸润的免疫细胞中表达增加[20-21]。IPF患者肺组织中OPN与MMP7共定位,OPN诱导MMP7的表达和活性,而OPN被MMP7切割和激活[19,22]。MMP7可以特异性切割核心蛋白聚糖,从而从复合物中释放转化生长因子-β[23]。IBD中MMP-7的异常升高与黏膜糜烂、结构组织改变和炎症浸润有关,MMP-7的过度表达有助于IBD的肠屏障损伤[24-25]。
与非IBD人群相比,IBD患者黏膜转化生长因子TGF-β水平升高[26-27]。COL1A1和COL3A1是细胞外基质相关基因,在IPF发病机制中发挥重要作用。IPF的特征是促纤维化细胞因子TGF-β1的上调,TGF-β1与包括胶原(主要是Ⅰ型和Ⅲ型)在内的ECM蛋白的产生增强相关[27]。COL3A1编码Ⅲ型胶原蛋白α1链,COL1A1编码Ⅰ型胶原蛋白α1[28]。COL1A1在IBD患者的炎症组织中表达增加[29]。由POSTN基因编码的骨膜蛋白主要通过PI3K/Akt和FAK途径与蛋白受体相互作用,导致组织重塑、纤维化、炎症等多种病理过程[30]。骨膜蛋白在IPF患者中表达增加,定位于活性纤维化区域[31]。TGF-β和IL-13可促进纤维化肺实质中骨膜蛋白的表达[32]。既往研究表明,骨膜蛋白激活NF-κB信号通路介导肠道炎症,提示骨膜蛋白是IBD的潜在治疗靶点[33]。
TFs在基因表达调控中发挥重要作用。在TFs-基因互作网络中,SPP1、MMP7和COL6A3与其他TFs基因的相互作用率较高。SPP1受10个TFs基因调控,MMP7和COL6A3受9个TFs基因调控。调控因子中,HINFP、NFYA、POU2F2、YY1、FOXL1和FOXC1是TFs-基因相互作用网络中程度最高的调控因子。HINFP、POU2F2、YY1、FOXL1和FOXC1均靶向SPP1。TFs和miRNAs均可调控靶基因的表达,且相互调控,在多种生物过程和疾病的分化中发挥重要作用[34]。通过TFs-miRNAs共调控网络,了解到核心基因、TFs和miRNAs之间的关系。在已鉴定的TFs中,JUN的级别最高,为4级。JUN蛋白包含c-Jun、JunB和JunD,其中c-Jun是最有效的转录激活因子,一项研究表明,IPF中异常的致病性成纤维细胞需要c-JUN来快速增殖[35-36]。在IBD患者中,严重受损组织中c-Jun基因表达水平高于外观正常组织[37]。在miRNAs中,hsa-miR-301b, hsa-miR-301a, hsa-miR-29c, hsa-miR-29b和hsa-miR-29a的表达度最高,程度为3,且均靶向COL6A3。有研究表明,COL6A3是TGF-β/Smad信号通路的靶点,可能是IPF的潜在生物标志物[38-39]。在IBD中,COL6A3编码的COL6α3链在CD和UC患者的肠组织中升高[40]。
4 结论
通过先进的生物信息学方法,建立了IPF和IBD的PPI网络,并筛选了SPP1、COL1A1、POSTN、MMP7、COL3A1、COL6A3等枢纽候选基因。同时,通过生物信息学分析,分析了分子调控网络中的信号通路。本研究发现了IPF和IBD的共同靶点和功能通路,并通过可视化网络图更清晰地表达了两者的相互作用。这些结果表明IPF与IBD之间存在相似之处和潜在的关系,可作为未来研究的理论基础,并为诊断和治疗提供新的潜在靶点。然而,此研究也存在一些局限性。首先,目前的研究只涉及6个核心基因。其次,TFs-基因和TFs-miRNAs-基因相互作用网络仅基于公共数据库的预测,缺乏对IPF和IBD中核心基因、TFs和miRNAs调控的分子机制的详细研究。因此,这些枢纽基因和miRNAs在IPF和IBD发生中的分子机制有待进一步研究。
