牦牛基因组拷贝数变异研究进展
2024-04-02徐东辉徐宇辉李瑞哲成海建马志杰
徐东辉,徐宇辉,李瑞哲,成海建,马志杰*
(1.青海大学畜牧兽医科学院,西宁 810016;2.农业农村部青藏高原畜禽遗传育种重点实验室,西宁 810016;3.青海省高原家畜遗传资源保护与创新利用重点实验室,西宁 810016;4.山东省农业科学院畜牧兽医研究所,济南 250100;5.山东省畜禽疫病防治与繁育重点实验室,济南 250100)
拷贝数变异(copy number variation, CNV)是指物种基因组中大片段DNA片段的插入、缺失或重复等,是基因组结构变异的主要形式之一。CNV是物种个体遗传变异的重要来源,在不同物种基因组上普遍存在。研究表明,基因组中拷贝数的分布受到基因的突变、自然选择以及种群数量的发展趋势等多个因素影响[1]。CNV能影响动物基因组中较长区域,甚至产生巨大的遗传效应,与基因组进化密切相关。如今,CNV的定义范围已扩大到了50 bp到数Mb序列长度的结构变异。与单核苷酸多态性(single nucleotide polymorphism,SNP)相比,CNV覆盖了物种更多的基因组区域,并拥有更高的核苷酸位点突变率。当前,CNV已成为继简单重复序列(simple sequence repeat, SSR)和SNP之后一种新的基因组变异类型而成为研究热点之一。
随着组学(omics)和高通量测序技术的发展,全基因组CNV研究在普通牛[2]、羊[3]、猪[4]等家畜中已陆续开展,研究证实一些CNV关联基因或基因组区域与家畜的脂肪沉积、免疫应答及环境适应性等相关,表明CNV分析是探究动物表型性状遗传机理和开展群体遗传等研究的重要策略之一。如Huang等[3]通过对普通牛、绵羊和山羊全基因组CNV分析,发现830个与上述反刍动物耐药性、免疫和肌肉发育等多种生命活动相关且在群体间高度分化(VST≥ 0.5)的CNV,在普通牛、山羊、绵羊中分别发现11、26、16个与环境适应性显著相关的CNV,提示种间共享的拷贝数变异区域(copy number variation region,CNVR)比例远高于种间共享的SNP,推测CNV热点(hotspots)对种间共享CNVR的形成影响很小,以平衡选择为主的自然选择对种间共享CNVR的形成和保留可能起到关键作用。
牦牛作为青藏高原的特有牛种,其全基因组CNV研究有助于挖掘与牦牛生长、肉乳、高海拔适应性等重要性状和生理功能相关的关键基因,给未来牦牛分子育种和遗传改良提供研究基础。当前,在牦牛基因组CNV研究中,自Zhang等[5]利用比较基因组杂交芯片(comparative genomic hybridization, CGH)对2头牦牛进行首次基因组CNV探究后,基于Bovine HD芯片和基因组重测序策略,研究者近10年来对牦牛全基因组拷贝数变异进行了较多的分析,其研究内容主要涉及了牦牛全基因组CNV图谱的构建[6-9]、部分牦牛品种(群体)间CNV及CNVR遗传差异揭示[8-13]以及适应性、经济性状等相关的CNV关联候选基因或基因组区域的发掘与检测等[5-14]。然而,当前尚未见有关牦牛基因组CNV研究进展的综述报道。鉴于此,本文对CNV的形成原理、作用机制、检测方法以及牦牛全基因组CNV和基因拷贝数变异研究最新进展进行综述,提出了当前研究中存在的若干问题,并对其发展前景进行了展望,以期为今后继续推进牦牛基因组遗传变异探究,发掘有用遗传信息,深入开展牦牛分子育种实践奠定基础。
1 基因组CNV形成原理、作用机制和检测方法
1.1 CNV形成原理
CNV作为一种重要的基因组结构变异类型,其多态性差异会导致同一物种的不同个体间特定基因组区域内基因拷贝数的不同,进而引起物种表型发生变化。当前,基因组CNV的形成机制主要包括以下4类[15]:非等位同源重组(non-allelic homologous recombination, NAHR)、非同源末端连接(non-homologous end joining, NHEJ)、复制叉延迟和模板转换(fork stalling and template switching, FoSTeS)及LINE-1(long interspersed nucleotide element-1, L1)介导的反转录转座(图1)。
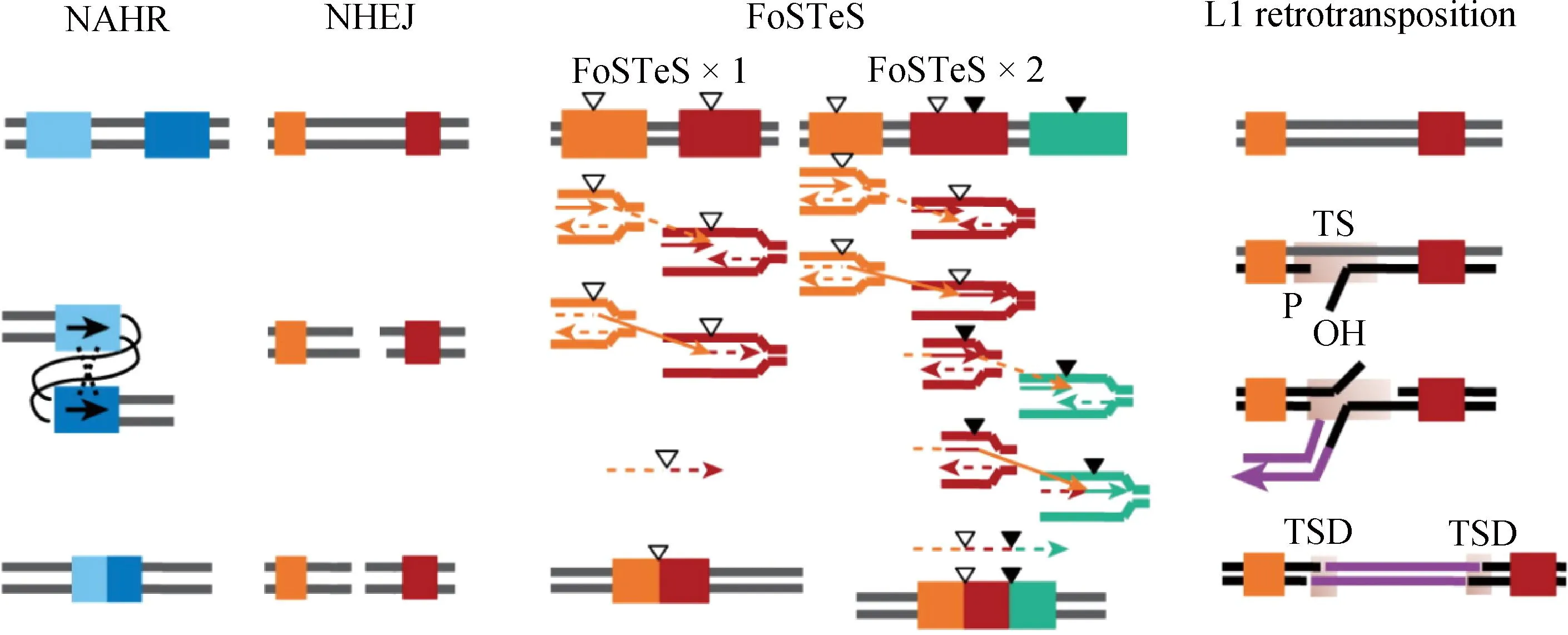
图1 基因组CNV四种形成机制[15]Fig.1 Four mechanisms of genomic CNV formation[15]
NAHR是在减数分裂或有丝分裂中,非同源染色体间序列相似的两个区域之间发生的重组。如果姐妹染色单体之间发生交叉,该过程会发生DNA片段的增加或丢失,进而导致染色体片段的扩增、缺失和倒位[16]。NAHR代表性变异类型有两类,即大片段重复序列(segmental duplications, SDs)和低拷贝重复序列(low copy repeats, LCRs),其重复片段之间相似性达到95%以上,通常长度大于10 kb,且较为复杂的SD和LCR序列可由自身形成。NHEJ是一种细胞修复的生理形式,是两个非同源DNA序列在末端连接,由电离辐射或活性氧引起的DNA双链断裂(DNA double-strand breaks,DSBs),从而导致染色体重排产生CNV。FoSTeS是一种基于DNA复制的机制,它可以解释复杂的基因组重排和CNV[17]。在DNA复制过程中,停滞的复制叉导致滞后链脱落,并在其它复制叉上进行DNA复制合成。新的模板链与原来复制叉中的模板链彼此靠近,模板转换的结果可以导致缺失或重复。L1介导的反转录转座过程由RNA聚合酶II转录而成的RNA中间体完成介导[18],其在转录和反转录的转座过程中会产生新的CNV,并插入到基因组中两端为一对重复序列的位置上。
1.2 CNV作用机制
CNV主要通过基因剂量效应(gene dosage)和位置效应(position effects)影响基因的表达和物种的表型变化。基因剂量效应指CNV的缺失或重复会改变基因的表达量,进而引起基因功能表达异常及表型发生变化。值得注意的是,基因剂量效应会直接改变所在基因的表达水平,如在对人类腓骨肌萎缩症(charcot-marie-tooth)神经病变的研究中,发现该病特异性地与剂量敏感基因外周髓鞘蛋白22(PMP22)的CNV密切相关[19]。在人类孤独症相关研究中,发现一些与神经突触形成及突触蛋白合成相关的基因位点,如SHANK3、轴突蛋白neurexin1和神经连接蛋白(neuroligin,NLGN4)等基因存在CNV的缺失[20]。位置效应则是指CNV对其变异位点周围基因的表达产生调控的间接影响,进而引起基因功能发生改变。
1.3 CNV检测方法
目前, CNV的检测可分为全基因组范围内未知CNV的检测和已探明CNV的检测与确定。全基因组范围内对未知CNV的检测主要分为两类:一类是芯片法,主要包括比较基因组杂交芯片(array-based comparative genomic hybridization, aCGH)和SNP芯片的运用;另一类是以测序为基础的新一代测序技术(next-generation sequencing, NGS)和单分子测序技术(single molecule sequencing)。对已探明CNV的检测与确定,常用的有荧光原位杂交(fluorescence in situ hybridization, FISH)和荧光定量PCR(real-time quantitative polymerase chain reaction, qPCR)技术检测两种方法[21]。
1.3.1 全基因组未知CNV的检测与确定 aCGH是基于不同的荧光标记,将对照样本和试验样本标记后与芯片上DNA片段杂交,通过检测不同的荧光信号和比值进行分析,来确定试验样本的拷贝数变化情况。1998年,Pinkel等[22]基于微阵列技术将不同荧光素标记的样本置于同一张芯片上,与构建的文库进行杂交,从而根据荧光信号值的不同在全基因组范围内检测CNV差异。CGH芯片的探针合成速度快,可搭载高密度探针,是一种高精度、高灵敏度、高分辨率且样本需要量较小的高通量分析技术。通常,探针的密度决定了结果的准确性。根据探针来源的不同,CGH芯片分为细菌人工染色体芯片和寡核苷酸芯片。其中,细菌人工染色体芯片具有较高的信噪比,但分辨率较低,很难检测到50 kb以下的CNV信号,同时制备该芯片的成本高,且费时费力。相比之下,寡核苷酸芯片则具有高精度、高分辨率、易制备等优点。与aCGH相比,高密度SNP芯片无需参照样品,其通过检测样本SNP强度来确定基因拷贝数变异。2007年,Affymetrix公司发布了全基因组SNP 6.0芯片,该芯片可将CNV转化成高分辨率的参考图谱,其包括90多万个用于拷贝数变化检测的探针,可使全基因组平均分辨率达3 kb,方便研究者通过CNV分析来挖掘基因组序列变异信息[23]。2008年,Kato等[24]利用芯片探针和最大似然法,构建隐马尔可夫模型并把检测出的芯片信号强度与参考杂交信号强度进行比较,推断人类基因组CNV区域上的复杂单倍型,得到了个体CNV区域内两条同源染色体上等位基因的基因拷贝数。高密度SNP芯片的问世,尽管大幅度提高了全基因组CNV的检测效率,但高密度SNP芯片对基因组扩增富集区和结构复杂区域内的CNVs检测效率仍然较低。
当前,对于全基因组测序数据,通常有4种不同的方法来检测其拷贝数变异:1)读段深度(read depth, RD):根据滑动窗口的标准化读取深度判断CNV;2)双末端映射(paired-end mapping, PEM):通过将paired-end间的距离与参考基因组比对来确认CNV;3)拆分reads (split reads, SR);4)reads组装(assembly, AS)[25-27]。其中,RD法在CNV检测中应用最为广泛。相较于aCGH和SNP芯片,NGS的有效分辨率和灵敏度更高,可用于确定基因组CNV的边界并能有效地检测小片段的CNV,在基因组断点的检测上有很大的优势。同时,得益于高通量测序成本的持续降低以及NGS检测CNV技术的不断成熟,高通量测序技术已被普遍应用于各个物种的CNV检测及相关研究中。此外,NGS技术能够更精准的覆盖基因组中片段重复(segmental duplications, SDs)和拷贝数变异区域(copy number variation regions, CNVRs),表现出前所未有的发掘CNVs的能力,所挖掘出的CNVs信息也极大丰富了芯片设计资源,推动了高精度芯片的持续升级。
1.3.2 已知CNV的检测与鉴定 FISH是将已知的带有荧光标记的单链核酸探针与目的DNA单链核酸片段进行杂交,利用荧光信号进行定性分析。该方法是基于形态基础的分子检测,具有检测速度快、信号强、能够多重染色的优势。然而,由于其技术复杂、成本高,应用短片段的cDNA探针时检测效率低,不能应用于候选基因或基因调控区内CNV的验证。qPCR是在聚合酶链式反应(PCR)中添加荧光标记内参基因,通过连续侦测荧光信号,统计每次PCR循环后产物总量,基于Ct值和标准曲线的关系对起始模板进行定量分析。qPCR通过对样本的目标基因与内参基因的检测值相对定量来推断目标基因的拷贝数[28],其操作简便、快速,灵敏度高,可实时监控且特异性更高,被研究者们视为CNV检测与鉴定的金标准。
2 牦牛基因组拷贝数变异(CNV)研究进展
2.1 全基因组CNV研究
牦牛全基因组CNV研究中,基于芯片技术,Zhang等[5]利用aCGH法对24头普通牛、3头水牛和2头牦牛进行了全基因组CNV分析,共确定了605个CNVRs,覆盖普通牛基因组的3.04%,研究发现41.8%(253/605)的CNVRs涉及与结合蛋白、受体活性及分子转导活性等相关的功能基因,70.6%(427/605)的CNVRs与数量性状基因座(quantitative trait loci, QTL)重叠;证实CNVRs在染色体上存在不均匀分布以及线粒体DNA拷贝数存在牛种间差异(其中扩增型:普通牛;缺失型:牦牛和水牛);qPCR进一步验证CNVR14对PLA2G2D基因表达呈显著负影响,而CNVR22和CNVR310与普通牛的体尺性状呈显著负相关。张全伟[6]使用普通牛Bovine HD芯片检测了200头牦牛(包括青海牦牛和天祝白牦牛)的全基因组CNV,共获得总长128 Mb的857个CNVRs(覆盖牦牛基因组的4.79%),功能富集分析发现大部分候选基因与嗅觉受体活性、细胞表面受体信号转导和G蛋白偶联受体信号通路相关;以普通牛基因组为参考,将获得的CNVRs定位到普通牛染色体上,初步构建了牦牛常染色体CNVRs遗传图谱。随后,Jia等[7]用Illumina Bovine HD芯片在215头阿什旦牦牛基因组中检测到了总长181.6 Mb的1 066个CNVRs(覆盖了普通牛常染色体基因组的7.2%),并获得了牦牛全基因组CNV图谱;功能富集揭示了许多潜在的与高海拔适应相关的基因,检测发现超过1/3的CNVRs与牦牛肉、奶、繁殖等重要经济性状相关的QTL重叠。
基于高通量测序技术,Zhang等[8]对14头野牦牛和65头家牦牛全基因组重测序数据(平均测序深度6.7×)进行了CNV扫描,确定了总长度为153 Mb的2 634个CNVRs,占牦牛参考基因组的5.7%;家、野牦牛的比较分析共确定出121个差异CNVRs,这些区域包含与繁殖、营养、神经发育和能量代谢相关的基因;而在高、低海拔家牦牛群体分组分析中,确定了85个呈显著差异的CNVRs,发现了与低氧应激(DEXI、DCC、MRP4)、免疫防御(ULBP17、CIITA、CATHLI1、BOLA-DQA2、BOLA-DQA3和BOLA-DQB)等相关的一些基因。Wang等[9]基于全基因组重测序数据(平均测序深度8.1×)对中国16个牦牛群体共48头牦牛个体进行了全基因组CNV分析,得到总长163.8 Mb的3 174个CNVRs(占牦牛参考基因组的6.2%),发现这些CNVRs包含与免疫反应、葡萄糖代谢、感觉感知及高海拔环境适应相关的功能基因(DCC、GSTCD、MRPS28和MOGAT2);聚类分析显示牦牛CNV分为2个来源,推测一些牦牛CNV可能在不同的群体中独立发生从而造成群体差异。此外,周学兰[10]对9头野牦牛、30头大通牦牛、30头青海高原牦牛和30头天祝白牦牛(平均测序深度27~30×)进行了全基因组CNV检测,共鉴定得到122 898个CNVs,发现大多数CNVs在各群体间共有,而野牦牛、大通牦牛、青海高原牦牛和天祝白牦牛特异性CNVs分别有17 833、17 127、15 027和16 878个;对群体特异性CNVs重叠基因进行功能富集分析,并与普通牛的QTL数据进行重叠分析,鉴定得到27个与牦牛经济性状、适应性和性情等相关的基因,包括与生长性状(WNT7A、COL1A1、ASB2、MECP2等)、繁殖性状(SPACA7、TAF7L和BMP15)、高原适应性(HCN4、FABP2、GADD45A、KRT71等)、性情(CACNA1H、GPR88、NLGN3、SEPT6等)、肉品质(ADIPOQ)相关的若干基因。E等[11]对14头西藏那曲市高海拔地区的牦牛个体和15头甘肃抓喜秀龙乡低海拔地区的牦牛个体重测序数据进行了全基因组拷贝数变异检测,从FST和VST差异最大的前20个CNVs中鉴定出7个候选CNVs,发现5个与牦牛适应性、生理功能调节相关的基因(即GRIK4、IFNLR1、LOC102275985、GRHL3和LOC102275713)。朱昌鸿[14]对3头麦洼牦牛、2头犏牛、9头普通牛共14头个体的全基因组重测序数据进行分析,共鉴定出普通牛、牦牛、犏牛特异性CNVs相关基因5 384、392和1 969个;在牦牛中筛选到与骨骼发育、生长性状相关的基因(DLx5、LYZL4等),在犏牛中筛选到与肉质、高原低氧适应性相关的若干基因(ADIPOQ、UPC1、AQP7等)。Meng等[12]对20头天祝白牦牛(包括10头长毛个体和10头正常毛长个体)的重测序数据进行了全基因组CNV检测,定义了2 006个CNVRs,发现80个差异CNVRs主要富集了与脂质代谢、细胞迁移等相关的基因,并检测到与毛发生长、毛囊发育相关的若干差异基因(ASTN2、ATM、COL22A1、GK5、SLIT3、PM20D1和SGCZ)。最近,Zhang等[13]基于38头纯白天祝白牦牛和59头非纯白牦牛个体探究了牦牛被毛白色表型的遗传机制,定义了两个群体间高度分化的拷贝数变异区域,并鉴定出KIT基因周围两种可能与牦牛被毛白色表型相关的CNVs类型(CNV1和CNV2),该研究结合全基因组SNP及GWAS结果发现,在白色牦牛中存在6号、29号染色体之间易位以及KIT基因连锁的Cs等位基因;与普通牛相比,白色牦牛的结构变异显示了Cs等位基因的额外重复,与牦牛白色被毛表型相关的Cs等位基因存在来自普通牛的基因渗入。
综上可以看出,研究者已基于高密度芯片和全基因组重测序技术从全基因组水平对我国部分牦牛品种(群体)进行了CNV检测和相应的品种(群体)间差异分析,丰富了牦牛CNV数据集(库),发现了一批与牦牛适应性、性情及重要经济性状(如生长发育、毛长、肉质)等密切相关的候选基因或CNVR,初步揭示了牦牛高海拔适应、驯化及毛色形成等的分子机制及其功能调控模式。尽管上述研究取得了一些初步成果,但仍存在些许不足,今后如下若干研究内容仍有待深入探究和推进:
1)牦牛基因组CNV研究中样本的代表性有待增强,样本量可再增加和提高。先前研究中牦牛品种(群体)样本覆盖率低,数量较少,全基因组CNV研究中各牦牛品种(群体)样本数目大多不足4头,如Zhang等[8]的研究中仅只涉及到4个省/区的牦牛样本,未覆盖到新疆与云南牦牛产区;而Wang等[9]的研究中涉及到16个牦牛群体,各群体分别只选择了3头个体。目前,我国牦牛品种(遗传资源)已达23个[29-32]。因此,今后可选择覆盖面更广、数目更多的样本,这对构建高质量牦牛CNV遗传图谱、丰富牦牛基因组变异数据库有重大意义。
2)牦牛全基因组测序深度有待提高,进而降低数据CNV检测出现的假阳性影响。当前,已报道的牦牛CNV研究基于全基因组测序获得的重测序数据测序深度大多不足10×[8-9]。随着测序技术的发展、测序成本的降低及三代测序技术的普及应用,利用二代测序高通量、准确度高的短读长片段对三代测序的长读长片段进行修正,有助于建立更为精准、完善的牦牛CNV研究新标准。
3)牦牛基因组CNV识别标准有待建立。CNV的识别是CNV研究中的核心,目前CNV的识别缺乏统一标准,其方法主要分为测序法和芯片法。aCGH和SNP芯片法假阳性较高,而测序法在一些CNV小片段和高度重复的复杂区域内识别亦存在较大缺陷。CNV识别受限于检测方法、灵敏度等因素,这使得不同软件、不同算法获得的结果不能进行有效的整合和比较分析,未来可在软件开发领域着手建立牦牛CNV的统一识别标准,来解决上述识别问题。
4)染色体组装水平的牦牛高质量参考基因组应在CNV检测中发挥作用。目前,大多牦牛CNV研究使用的参考基因组为BosGru_v2.0[7-9,33],其仅组装到Scaffold水平,而报道的牦牛染色体水平的CNV图谱研究也是基于普通牛参考基因组绘制获得[6],其结果缺乏准确性。2021年以来,牦牛全基因组序列已被组装到了染色体水平[34-35](即BosGru3.1,NWIPB_DYAK_1.0和NWIPB_WYAK_1.0)。受限于Contig、Scaffold水平的参考基因组中存在大量的gap,故选用染色体水平的高质量参考基因组对深度解析牦牛CNV具有重要作用。
2.2 基因CNV研究
牦牛基因拷贝数变异(CNV)研究主要是探究一些候选基因的不同CNV状态及与重要经济性状、生理功能的关联情况,分析其调控作用并进行功能验证。基因拷贝数差异在不同牦牛品种(群体)间普遍存在,对性状具有一定的影响。当前,研究者已在10个牦牛品种(即大通、甘南、青海高原、阿什旦、天祝、麦洼、金川、木里、昌台和玉树牦牛)中开展了候选基因CNV与部分性状的关联性分析(表1),初步证实部分基因(如HPGDS、KLF6、CHRM3等)的CNV类型与一些肉质、生长性状存在关联,其研究成果为揭示牦牛性状的遗传机理奠定了基础,对加快牦牛分子育种进程有重要意义。

表1 牦牛基因拷贝数变异研究
然而,上述研究仍存在若干问题,如样本的代表性不强、候选基因CNV的验证研究较少等。今后,牦牛候选基因的CNV与部分性状的关联性验证有待继续推进。先前研究中挖掘出的一批与牦牛重要性状相关联的候选基因,尚需对其进行检测和验证以明确该基因的拷贝数状态。另一方面,CNV全基因组上的分布在不同群体、个体间存在差异性,因此,在对已知CNV验证时应选择多个牦牛群体的大样本量开展相应研究。
3 展 望
可以看出,当前研究者在牦牛全基因组拷贝数变异和基因拷贝数变异研究中已开展了大量工作,其研究成果可概括为:1)在牦牛全基因组首次测序完成后[33],构建了Scaffold水平的牦牛全基因组CNV图谱[7-9],并初步构建了基于普通牛参考基因组的牦牛染色体水平的CNV图谱[6]。2)对牦牛全基因组进行检测,挖掘出大量牦牛CNVs信息,鉴定出一批与适应性、生长性状、免疫等相关的牦牛CNVRs及CNVRs上的候选基因。3)分析了部分牦牛品种(群体)间(包括不同海拔梯度分组群间以及不同表型(毛长)群体间)差异CNVRs,将其与SDs、QTL的重叠区域相关联,注释得到一批相关的候选基因,为揭示牦牛品种(群体)间遗传差异提供了基础数据。4)对牦牛部分基因的CNV与生长、繁殖、肉质等性状的关联性进行了验证,探究了牦牛和犏牛Y染色体雄性特异区多拷贝基因的CNV状态及其对犏牛雄性不育的影响。
当前,牦牛基因组CNV研究仍处于发展阶段。由于基因组CNV结构的复杂性和相应的基因组CNV检测技术存在的局限性,CNV检测错误率较高以及一些复杂基因组区域无法覆盖成为当前亟待解决的问题。为实现CNV的准确鉴定,可组合使用不同检测方法的优势,并结合三代长片段测序和机器学习的方法有效降低检测的假阳性率。整合不同来源的CNV数据,开发新的预测模型及高精度芯片对准确识别CNV、解决基因大批量精确分型具有重要意义。同时,随着泛基因组、单细胞多组学的快速发展,单细胞CNV分析和空间转录组学技术已被应用于推断人体组织的空间拷贝数变异状况[51],这为后续开展牦牛空间基因组拷贝数变异分析和探究更复杂基因组区域拷贝数状态提供了借鉴。此外,随着三代测序技术的发展,应用PacBio HiFi、ONT ultra-long、Hi-C等技术已经在人[52-55]、水稻[56]、玉米[57]等动植物上实现了高质量的端粒到端粒的基因组组装(telomere-to-telomere, T2T),大幅提高了染色体的连续性和完整性。可以预见,未来牦牛T2T基因组的组装也会很快提上日程,这将极大促进牦牛全基因组CNVs的深入挖掘及其生物学功能的探索,对开展牦牛分子育种实践将具有重要意义。
在育种方面,结合基因组CNV、SV及SNP等遗传变异开展GWAS研究,来选择与表型特征、抗病、重要经济性状等相关的候选基因,构建统计模型将其整合到全基因组选择中,是未来牦牛分子育种工作的重心之一。随着基因组测序技术与CNV新算法的不断发展,结合以CRISPR/Cas9系统为核心的基因组编辑技术,将有助于开发与牦牛优良性状相关的CNV分子标记,提供更高效、安全的遗传标记来应用到牦牛育种中。可以相信,深入开展CNV研究并将其作为牦牛育种中的主要分子标记之一,针对优良性状进行品种选育,将有力推动牦牛遗传改良和分子育种进程。
